Working Group 1 - Chapter 2: Changes in Atmospheric Constituents and in Radiative Forcing - (AR4-WG1-2)
Original at: http://www.ipcc.ch/publications_and_data/ar4/wg1/en/ch2.html
Main AR4 Index | Working Group WG1 Index | Table of Contents | Authors | Executive Summary | Annotated Text | References | Reviewer Comments
With the exception of Chapter and Section headings, all coloured text has been inserted by AccessIPCC. The non-coloured text is the IPCC original.
A number of emails from the Climate Research Unit (CRU) of the University of East Anglia were published on the Internet in November 2009. This has provided a window into the world of climate science.
We have identified a number of key individuals involved in the emails whom we have designated as Persons of Concern [PoC]; a Journal in which a PoC has published has been designated as a Journal of Concern [JoC].
This is not to suggest that we believe such papers are necessarily flawed, but rather that, as Joseph Alcamo noted at Bali in October 2009, "as policymakers and the public begin to grasp the multi-billion dollar price tag for mitigating and adapting to climate change, we should expect a sharper questioning of the science behind climate policy".
References occur in a list at the end of each chapter. Citations are within the normal text of sections and paragraphs.
| Tag | Explanation | Where Used | References | Citations |
|---|---|---|---|---|
| PoC |
Person of Concern Key individual involved in CRU emails as defined in this spreadsheet. |
References, Citations, IPCC Roles | 19 | 32 |
| JoC |
Journal of Concern A Journal which has published articles by one or more PoCs (Person of Concern) |
References, Citations | 585 | 964 |
| MoS |
Model or Simulation Reference appears to be a model or simulation, not observation or experiment |
References, Citations | 262 | 543 |
| NPR |
Non Peer Reviewed Reference has no Journal or no Volume or no Pages or it has Editors. |
References, Citations | 54 | 144 |
| SRC |
Self Reference Concern Author of a chapter containing references to own work. |
References, Citations, IPCC Roles | 165 | 381 |
| ARC |
Paper authored or co-authored by person who is also in list of Authors of another chapter. |
References, Citations | 113 | 279 |
| 2007 |
Paper dated 2007, when IPCC policy stated cutoff was December 2005 |
References, Citations | 6 | 11 |
| Ambiguous |
The short inline citation matched with more than one reference; however, AccessIPCC will link to the first reference found. |
Citations | - | 11 |
| NotFound |
The short inline citation was not matched with any reference. Believed to be caused by typing errors. |
Citations | - | 2 |
| Clean |
The reference was probably peer reviewed. |
References, Citations | 68 | 77 |
Coordinating Lead Authors:
Piers Forster (UK) [SRC:7], Venkatachalam Ramaswamy (USA) [SRC:11],
| Concern | Occurrence |
|---|---|
| SRC >= 5 | 2 |
| Potentially Biased Authors | 2 |
Lead Authors:
Paulo Artaxo (Brazil), Terje Berntsen (Norway) [SRC:4], Richard Betts (UK) [SRC:6], David W. Fahey (USA), James Haywood (UK) [SRC:13], Judith Lean (USA) [SRC:8], David C. Lowe (New Zealand) [SRC:6], Gunnar Myhre (Norway) [SRC:11], John Nganga (Kenya), Ronald Prinn (USA; New Zealand) [SRC:10], Graciela Raga (Mexico; Argentina), Michael Schulz (France; Germany) [SRC:5], Robert Van Dorland (Netherlands) [SRC:1],
| Concern | Occurrence |
|---|---|
| SRC >= 5 | 7 |
| SRC 1-4 | 2 |
| Potentially Biased Authors | 9 |
| Impartial Authors | 4 |
Contributing Authors:
G. Bodeker (New Zealand) [SRC:1], O. Boucher (UK; France) [SRC:17], W.D. Collins (USA) [SRC:2], T.J. Conway (USA) [SRC:1], E. Dlugokencky (USA) [SRC:5], J.W. Elkins (USA), D. Etheridge (Australia) [SRC:2], P. Foukal (USA) [SRC:1], P. Fraser (Australia), M. Geller (USA) [SRC:2], F. Joos (Switzerland) [SRC:2][PoC], , C.D. Keeling (USA) [SRC:2], R. Keeling (USA) [SRC:3], S. Kinne (Germany) [SRC:3], K. Lassey (New Zealand), U. Lohmann (Switzerland) [SRC:11], A.C. Manning (UK; New Zealand) [SRC:2], S. Montzka (USA) [SRC:2], D. Oram (UK) [SRC:2], K. O’Shaughnessy (New Zealand), S. Piper (USA) [SRC:1], G.-K. Plattner (Switzerland), M. Ponater (Germany) [SRC:8], N. Ramankutty (USA; India) [SRC:2], G. Reid (USA), D. Rind (USA) [SRC:3], K. Rosenlof (USA), R. Sausen (Germany) [SRC:10], D. Schwarzkopf (USA), S.K. Solanki (Germany; Switzerland) [SRC:2], G. Stenchikov (USA) [SRC:7], N. Stuber (UK; Germany) [SRC:4], T. Takemura (Japan) [SRC:3], C. Textor (France; Germany) [SRC:2], R. Wang (USA), R. Weiss (USA) [SRC:2], T. Whorf (USA) [SRC:2],
| Concern | Occurrence |
|---|---|
| PoC | 1 |
| SRC >= 5 | 6 |
| SRC 1-4 | 22 |
| Potentially Biased Authors | 28 |
| Impartial Authors | 9 |
Review Editors:
Teruyuki Nakajima (Japan) [SRC:5], Veerabhadran Ramanathan (USA) [SRC:4],
| Concern | Occurrence |
|---|---|
| SRC >= 5 | 1 |
| SRC 1-4 | 1 |
| Potentially Biased Authors | 2 |
This chapter should be cited as:
Forster, P., V. Ramaswamy, P. Artaxo, T. Berntsen, R. Betts, D.W. Fahey, J. Haywood, J. Lean, D.C. Lowe, G. Myhre, J. Nganga, R. Prinn, G. Raga, M. Schulz and R. Van Dorland, 2007: Changes in Atmospheric Constituents and in Radiative Forcing. In: Climate Change 2007: The Physical Science Basis. Contribution of Working Group I to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change [Solomon, S., D. Qin, M. Manning, Z. Chen, M. Marquis, K.B. Averyt, M.Tignor and H.L. Miller (eds.)]. Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA.
Executive Summary
Radiative forcing (RF) [1] is a concept used for quantitative comparisons of the strength of different human and natural agents in causing climate change. Climate model studies since the Working Group I Third Assessment Report (TAR; IPCC, 2001 [NPR] ) give medium confidence that the equilibrium global mean temperature response to a given RF is approximately the same (to within 25%) for most drivers of climate change.
For the first time, the combined RF for all anthropogenic agents is derived. Estimates are also made for the first time of the separate RF components associated with the emissions of each agent.
The combined anthropogenic RF is estimated to be +1.6 [–1.0, +0.8] [2] Wm–2,indicating that, since 1750, it is extremely likely [3] that humans have exerted a substantial warming influence on climate. This RF estimate is likely to be at least five times greater than that due to solar irradiance changes. For the period 1950 to 2005, it is exceptionally unlikely that the combined natural RF (solar irradiance plus volcanic aerosol) has had a warming influence comparable to that of the combined anthropogenic RF.
Increasing concentrations of the long-lived greenhouse gases (carbon dioxide (CO2), methane (CH4), nitrous oxide (N2O), halocarbons and sulphur hexafluoride (SF6); hereinafter LLGHGs) have led to a combined RF of +2.63 [±0.26] Wm–2.Their RF has a high level of scientific understanding. [4] The 9% increase in this RF since the TAR is the result of concentration changes since 1998 .
— The global mean concentration of CO2 in 2005 was 379 ppm, leading to an RF of +1.66 [±0.17] Wm–2.Past emissions of fossil fuels and cement production have likely contributed about three-quarters of the current RF, with the remainder caused by land use changes. For the 1995 to 2005 decade, the growth rate of CO2 in the atmosphere was 1.9 ppm yr–1 and the CO2 RF increased by 20%: this is the largest change observed or inferred for any decade in at least the last 200 years. From 1999 to 2005, global emissions from fossil fuel and cement production increased at a rate of roughly 3% yr–1.
— The global mean concentration of CH4 in 2005 was 1,774 ppb, contributing an RF of +0.48 [±0.05] Wm–2.Over the past two decades, CH4 growth rates in the atmosphere have generally decreased. The cause of this is not well understood. However, this decrease and the negligible long-term change in its main sink (the hydroxyl radical OH) imply that total CH4 emissions are not increasing.
— The Montreal Protocol gases (chlorofluorocarbons (CFCs), hydrochlorofluorocarbons (HCFCs), and chlorocarbons) as a group contributed +0.32 [±0.03] Wm–2 to the RF in 2005 . Their RF peaked in 2003 and is now beginning to decline.
— Nitrous oxide continues to rise approximately linearly (0.26% yr–1)and reached a concentration of 319 ppb in 2005, contributing an RF of +0.16 [±0.02] Wm–2.Recent studies reinforce the large role of emissions from tropical regions in influencing the observed spatial concentration gradients.
— Concentrations of many of the fluorine-containing Kyoto Protocol gases (hydrofluorocarbons (HFCs), perfluorocarbons, SF6)have increased by large factors (between 4.3 and 1.3) between 1998 and 2005 . Their total RF in 2005 was +0.017 [±0.002] Wm–2 and is rapidly increasing by roughly 10% yr–1.
— The reactive gas, OH, is a key chemical species that influences the lifetimes and thus RF values of CH4,HFCs, HCFCs and ozone; it also plays an important role in the formation of sulphate, nitrate and some organic aerosol species. Estimates of the global average OH concentration have shown no detectable net change between 1979 and 2004 .
Based on newer and better chemical transport models than were available for the TAR, the RF from increases in tropospheric ozone is estimated to be +0.35 [–0.1, +0.3] Wm–2,with a medium level of scientific understanding. There are indications of significant upward trends at low latitudes.
The trend of greater and greater depletion of global stratospheric ozone observed during the 1980 s and 1990 s is no longer occurring; however, it is not yet clear whether these recent changes are indicative of ozone recovery. The RF is largely due to the destruction of stratospheric ozone by the Montreal Protocol gases and it is re-evaluated to be –0.05 [±0.10] Wm–2,with a medium level of scientific understanding.
Based on chemical transport model studies, the RF from the increase in stratospheric water vapour due to oxidation of CH4 is estimated to be +0.07 [± 0.05] Wm–2,with a low level of scientific understanding. Other potential human causes of water vapour increase that could contribute an RF are poorly understood.
The total direct aerosol RF as derived from models and observations is estimated to be –0.5 [±0.4] Wm–2,with a medium-low level of scientific understanding. The RF due to the cloud albedo effect (also referred to as first indirect or Twomey effect), in the context of liquid water clouds, is estimated to be –0.7 [–1.1, +0.4] Wm–2,with a low level of scientific understanding.
— Atmospheric models have improved and many now represent all aerosol components of significance. Improved in situ,satellite and surface-based measurements have enabled verification of global aerosol models. The best estimate and uncertainty range of the total direct aerosol RF are based on a combination of modelling studies and observations.
— The direct RF of the individual aerosol species is less certain than the total direct aerosol RF. The estimates are: sulphate, –0.4 [±0.2] Wm–2;fossil fuel organic carbon, –0.05 [±0.05] Wm–2;fossil fuel black carbon, +0.2 [±0.15] Wm–2;biomass burning, +0.03 [±0.12] Wm–2;nitrate, –0.1 [±0.1] Wm–2;and mineral dust, –0.1 [±0.2] Wm–2.For biomass burning, the estimate is strongly influenced by aerosol overlying clouds. For the first time best estimates are given for nitrate and mineral dust aerosols.
— Incorporation of more aerosol species and improved treatment of aerosol-cloud interactions allow a best estimate of the cloud albedo effect. However, the uncertainty remains large. Model studies including more aerosol species or constrained by satellite observations tend to yield a relatively weaker RF. Other aspects of aerosol-cloud interactions (e.g., cloud lifetime, semi-direct effect) are not considered to be an RF (see Chapter7 ).
Land cover changes, largely due to net deforestation, have increased the surface albedo giving an RF of –0.2 [±0.2] Wm–2,with a medium-low level of scientific understanding. Black carbon aerosol deposited on snow has reduced the surface albedo, producing an associated RF of +0.1 [±0.1] Wm–2,with a low level of scientific understanding. Other surface property changes can affect climate through processes that cannot be quantified by RF; these have a very low level of scientific understanding.
Persistent linear contrails from aviation contribute an RF of +0.01 [–0.007, +0.02] Wm–2,with a low level of scientific understanding; the best estimate is smaller than in the TAR. No best estimates are available for the net forcing from spreading contrails and their effects on cirrus cloudiness.
The direct RF due to increases in solar irradiance since 1750 is estimated to be +0.12 [–0.06, +0.18] Wm–2,with a low level of scientific understanding. This RF is less than half of the TAR estimate.
— The smaller RF is due to a re-evaluation of the long-term change in solar irradiance, namely a smaller increase from the Maunder Minimum to the present. However, uncertainties in the RF remain large. The total solar irradiance, monitored from space for the last three decades, reveals a well-established cycle of 0.08% (cycle minimum to maximum) with no significant trend at cycle minima.
— Changes (order of a few percent) in globally averaged column ozone forced by the solar ultraviolet irradiance 11-year cycle are now better understood, but ozone profile changes are less certain. Empirical associations between solar-modulated cosmic ray ionization of the atmosphere and globally averaged low-level cloud cover remain ambiguous.
The global stratospheric aerosol concentrations in 2005 were at their lowest values since satellite measurements began in about 1980 . This can be attributed to the absence of significant explosive volcanic eruptions since Mt. Pinatubo in 1991 . Aerosols from such episodic volcanic events exert a transitory negative RF; however, there is limited knowledge of the RF associated with eruptions prior to Mt. Pinatubo.
The spatial patterns of RFs for non-LLGHGs (ozone, aerosol direct and cloud albedo effects, and land use changes) have considerable uncertainties, in contrast to the relatively high confidence in that of the LLGHGs. The Southern Hemisphere net positive RF very likely exceeds that in Northern Hemisphere because of smaller aerosol contributions in the Southern Hemisphere. The RF spatial pattern is not indicative of the pattern of climate response.
The total global mean surface forcing [5] is very likely negative. By reducing the shortwave radiative flux at the surface, increases in stratospheric and tropospheric aerosols are principally responsible for the negative surface forcing. This is in contrast to LLGHG increases, which are the principal contributors to the total positive anthropogenic RF.
2.1 Introduction and Scope
This chapter updates information taken from Chapters 3 to 6 of the IPCC Working Group I Third Assessment Report (TAR; IPCC, 2001 [NPR] ). It concerns itself with trends in forcing agents and their precursors since 1750, and estimates their contribution to the radiative forcing (RF) of the climate system. Discussion of the understanding of atmospheric composition changes is limited to explaining the trends in forcing agents and their precursors. Areas where significant developments have occurred since the TAR are highlighted. The chapter draws on various assessments since the TAR, in particular the 2002 World Meteorological Organization (WMO)-United Nations Environment Programme (UNEP) Scientific Assessment of Ozone Depletion ( WMO, 2003 [NPR] ) and the IPCC-Technology and Economic Assessment Panel (TEAP) special report on Safeguarding the Ozone Layer and the Global Climate System (IPCC/TEAP, 2005 ).
The chapter assesses anthropogenic greenhouse gas changes, aerosol changes and their impact on clouds, aviation-induced contrails and cirrus changes, surface albedo changes and natural solar and volcanic mechanisms. The chapter reassesses the ‘radiative forcing’ concept (Sections 2.2 and 2.8 ), presents spatial and temporal patterns of RF, and examines the radiative energy budget changes at the surface.
For the long-lived greenhouse gases (carbon dioxide (CO2), methane (CH4), nitrous oxide (N2O), chlorofluoro-carbons (CFCs), hydrochlorofluorocarbons (HCFCs), hydrofluorocarbons (HFCs), perfluorocarbons (PFCs) and sulphur hexafluoride (SF6), hereinafter collectively referred to as the LLGHGs; Section 2.3 ), the chapter makes use of new global measurement capabilities and combines long-term measurements from various networks to update trends through 2005 . Compared to other RF agents, these trends are considerably better quantified; because of this, the chapter does not devote as much space to them as previous assessments (although the processes involved and the related budgets are further discussed in Sections 7.3 and 7.4 ). Nevertheless, LLGHGs remain the largest and most important driver of climate change, and evaluation of their trends is one of the fundamental tasks of both this chapter and this assessment.
The chapter considers only ‘forward calculation’ methods of estimating RF. These rely on observations and/or modelling of the relevant forcing agent. Since the TAR, several studies have attempted to constrain aspects of RF using ‘inverse calculation’ methods. In particular, attempts have been made to constrain the aerosol RF using knowledge of the temporal and/or spatial evolution of several aspects of climate. These include temperatures over the last 100 years, other RFs, climate response and ocean heat uptake. These methods depend on an understanding of – and sufficiently small uncertainties in – other aspects of climate change and are consequently discussed in the detection and attribution chapter (see Section 9.2 ).
Other discussions of atmospheric composition changes and their associated feedbacks are presented in Chapter7 .Radiative forcing and atmospheric composition changes before 1750 are discussed in Chapter6 .Future RF scenarios that were presented in Ramaswamy et al. 2001 [NPR, MoS, SRC] ) are not updated in this report; however, they are briefly discussed in Chapter 10 .
2.2 Concept of Radiative Forcing
The definition of RF from the TAR and earlier IPCC assessment reports is retained. Ramaswamy et al. 2001 [NPR, MoS, SRC] ) define it as ‘the change in net (down minus up) irradiance (solar plus longwave; in W m–2) at the tropopause after allowing for stratospheric temperatures to readjust to radiative equilibrium, but with surface and tropospheric temperatures and state held fixed at the unperturbed values’. Radiative forcing is used to assess and compare the anthropogenic and natural drivers of climate change. The concept arose from early studies of the climate response to changes in solar insolation and CO2,using simple radiative-convective models. However, it has proven to be particularly applicable for the assessment of the climate impact of LLGHGs ( Ramaswamy et al., 2001 [NPR, MoS, SRC] ). Radiative forcing can be related through a linear relationship to the global mean equilibrium temperature change at the surface (ΔTs): ΔTs = λRF, where λ is the climate sensitivity parameter (e.g., Ramaswamy et al., 2001 [NPR, MoS, SRC] ). This equation, developed from these early climate studies, represents a linear view of global mean climate change between two equilibrium climate states. Radiative forcing is a simple measure for both quantifying and ranking the many different influences on climate change; it provides a limited measure of climate change as it does not attempt to represent the overall climate response. However, as climate sensitivity and other aspects of the climate response to external forcings remain inadequately quantified, it has the advantage of being more readily calculable and comparable than estimates of the climate response. Figure 2.1 shows how the RF concept fits within a general understanding of climate change comprised of ‘forcing’ and ‘response’. This chapter also uses the term ‘surface forcing’ to refer to the instantaneous perturbation of the surface radiative balance by a forcing agent. Surface forcing has quite different properties than RF and should not be used to compare forcing agents (see Section 2.8.1 ). Nevertheless, it is a useful diagnostic, particularly for aerosols (see Sections 2.4 and 2.9 ).
Since the TAR a number of studies have investigated the relationship between RF and climate response, assessing the limitations of the RF concept; related to this there has been considerable debate whether some climate change drivers are better considered as a ‘forcing’ or a ‘response’ ( Hansen et al., 2005 [JoC, MoS, ARC] ; Jacob et al., 2005 [NPR, MoS, ARC] ; Section 2.8 ). Emissions of forcing agents, such as LLGHGs, aerosols and aerosol precursors, ozone precursors and ozone-depleting substances, are the more fundamental drivers of climate change and these emissions can be used in state-of-the-art climate models to interactively evolve forcing agent fields along with their associated climate change. In such models, some ‘response’ is necessary to evaluate the RF. This ‘response’ is most significant for aerosol-related cloud changes, where the tropospheric state needs to change significantly in order to create a radiative perturbation of the climate system ( Jacob et al., 2005 [NPR, MoS, ARC] ).
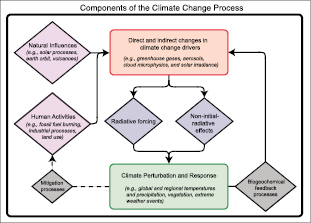
Figure 2.1. Diagram illustrating how RF is linked to other aspects of climate change assessed by the IPCC. Human activities and natural processes cause direct and indirect changes in climate change drivers. In general, these changes result in specific RF changes, either positive or negative, and cause some non-initial radiative effects, such as changes in evaporation. Radiative forcing and non-initial radiative effects lead to climate perturbations and responses as discussed in Chapters 6 , 7 and 8 .Attribution of climate change to natural and anthropogenic factors is discussed in Chapter9 .The coupling among biogeochemical processes leads to feedbacks from climate change to its drivers( Chapter7 ). An example of this is the change in wetland emissions of CH4 that may occur in a warmer climate. The potential approaches to mitigating climate change by altering human activities (dashed lines) are topics addressed by IPCC’s Working Group III.
Over the palaeoclimate time scales that are discussed in Chapter6 ,long-term changes in forcing agents arise due to so-called ‘boundary condition’ changes to the Earth’s climate system (such as changes in orbital parameters, ice sheets and continents). For the purposes of this chapter, these ‘boundary conditions’ are assumed to be invariant and forcing agent changes are considered to be external to the climate system. The natural RFs considered are solar changes and volcanoes; the other RF agents are all attributed to humans. For the LLGHGs it is appropriate to assume that forcing agent concentrations have not been significantly altered by biogeochemical responses (see Sections 7.3 and 7.4 ), and RF is typically calculated in off-line radiative transfer schemes, using observed changes in concentration (i.e., humans are considered solely responsible for their increase). For the other climate change drivers, RF is often estimated using general circulation model (GCM) data employing a variety of methodologies ( Ramaswamy et al., 2001 [NPR, MoS, SRC] ; Stuber et al., 2001b [MoS, SRC] ; Tett et al., 2002 [JoC, MoS, ARC] ; Shine et al., 2003 [JoC, MoS] ; Hansen et al., 2005 [JoC, MoS, ARC] ; Section 2.8.3 ). Often, alternative RF calculation methodologies that do not directly follow the TAR definition of a stratospheric-adjusted RF are used; the most important ones are illustrated in Figure 2.2 .For most aerosol constituents (see Section 2.4 ), stratospheric adjustment has little effect on the RF, and the instantaneous RF at either the top of the atmosphere or the tropopause can be substituted. For the climate change drivers discussed in Sections 7.5 and 2.5 ,that are not initially radiative in nature, an RF-like quantity can be evaluated by allowing the tropospheric state to change: this is the zero-surface-temperature-change RF in Figure 2.2 (see Shine et al., 2003 [JoC, MoS] ; Hansen et al., 2005 [JoC, MoS, ARC] ; Section 2.8.3 ). Other water vapour and cloud changes are considered climate feedbacks and are evaluated in Section 8.6 .
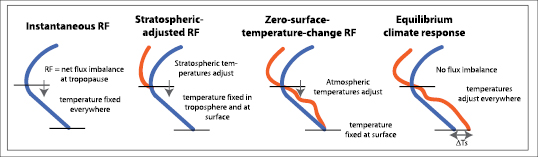
Figure 2.2. Schematic comparing RF calculation methodologies. Radiative forcing, defined as the net flux imbalance at the tropopause, is shown by an arrow. The horizontal lines represent the surface (lower line) and tropopause (upper line). The unperturbed temperature profile is shown as the blue line and the perturbed temperature profile as the red line. From left to right: Instantaneous RF: atmospheric temperatures are fixed everywhere; stratospheric-adjusted RF: allows stratospheric temperatures to adjust; zero-surface-temperature-change RF: allows atmospheric temperatures to adjust everywhere with surface temperatures fixed; and equilibrium climate response: allows the atmospheric and surface temperatures to adjust to reach equilibrium (no tropopause flux imbalance), giving a surface temperature change (ΔTs).
Climate change agents that require changes in the tropospheric state (temperature and/or water vapour amounts) prior to causing a radiative perturbation are aerosol-cloud lifetime effects, aerosol semi-direct effects and some surface change effects. They need to be accounted for when evaluating the overall effect of humans on climate and their radiative effects as discussed in Sections 7.2 and 7.5 .However, in both this chapter and the Fourth Assessment Report they are not considered to be RFs, although the RF definition could be altered to accommodate them. Reasons for this are twofold and concern the need to be simple and pragmatic. Firstly, many GCMs have some representation of these effects inherent in their climate response and evaluation of variation in climate sensitivity between mechanisms already accounts for them (see ‘efficacy’, Section 2.8.5 ). Secondly, the evaluation of these tropospheric state changes rely on some of the most uncertain aspects of a climate model’s response (e.g., the hydrologic cycle); their radiative effects are very climate-model dependent and such a dependence is what the RF concept was designed to avoid. In practice these effects can also be excluded on practical grounds – they are simply too uncertain to be adequately quantified (see Sections 7.5 , 2.4.5 and 2.5.6 ).
The RF relationship to transient climate change is not straightforward. To evaluate the overall climate response associated with a forcing agent, its temporal evolution and its spatial and vertical structure need to be taken into account. Further, RF alone cannot be used to assess the potential climate change associated with emissions, as it does not take into account the different atmospheric lifetimes of the forcing agents. Global Warming Potentials (GWPs) are one way to assess these emissions. They compare the integrated RF over a specified period (e.g., 100 years) from a unit mass pulse emission relative to CO2 (see Section 2.10 ).
2.3 Chemically and Radiatively Important Gases
2.3.1 Atmospheric Carbon Dioxide
This section discusses the instrumental measurements of CO2,documenting recent changes in atmospheric mixing ratios needed for the RF calculations presented later in the section. In addition, it provides data for the pre-industrial levels of CO2 required as the accepted reference level for the RF calculations. For dates before about 1950 indirect measurements are relied upon. For these periods, levels of atmospheric CO2 are usually determined from analyses of air bubbles trapped in polar ice cores. These time periods are primarily considered in Chapter6 .
Awide range of direct and indirect measurements confirm that the atmospheric mixing ratio of CO2 has increased globally by about 100 ppm (36%) over the last 250 years, from a range of 275 to 285 ppm in the pre-industrial era (AD 1000 – 1750 ) to 379 ppm in 2005 (see FAQ 2.1 ,Figure 1). During this period, the absolute growth rate of CO2 in the atmosphere increased substantially: the first 50 ppm increase above the pre-industrial value was reached in the 1970 s after more than 200 years, whereas the second 50 ppm was achieved in about 30 years. In the 10 years from 1995 to 2005 atmospheric CO2 increased by about 19 ppm; the highest average growth rate recorded for any decade since direct atmospheric CO2 measurements began in the 1950 s. The average rate of increase in CO2 determined by these direct instrumental measurements over the period 1960 to 2005 is 1.4 ppm yr-1.
High-precision measurements of atmospheric CO2 are essential to the understanding of the carbon cycle budgets discussed in Section 7.3 .The first in situ continuous measurements of atmospheric CO2 made by a high-precision non-dispersive infrared gas analyser were implemented by C.D. Keeling from the Scripps Institution of Oceanography (SIO) (see Section 1.3 ). These began in 1958 at Mauna Loa, Hawaii, located at 19°N ( Keeling et al., 1995 [JoC, SRC] ). The data documented for the first time that not only was CO2 increasing in the atmosphere, but also that it was modulated by cycles caused by seasonal changes in photosynthesis in the terrestrial biosphere. These measurements were followed by continuous in situ analysis programmes at other sites in both hemispheres ( Conway et al., 1994 [JoC, SRC] ; Nakazawa et al., 1997 [JoC] ; Langenfelds et al., 2002 [JoC] ). In Figure 2.3 ,atmospheric CO2 mixing ratio data at Mauna Loa in the Northern Hemisphere (NH) are shown with contemporaneous measurements at Baring Head, New Zealand in the Southern Hemisphere (SH; Manning et al., 1997 [NPR, ARC] ; Keeling and Whorf, 2005 [NPR, SRC] ). These two stations provide the longest continuous records of atmospheric CO2 in the NH and SH, respectively. Remote sites such as Mauna Loa, Baring Head, Cape Grim (Tasmania) and the South Pole were chosen because air sampled at such locations shows little short-term variation caused by local sources and sinks of CO2 and provided the first data from which the global increase of atmospheric CO2 was documented. Because CO2 is a LLGHG and well mixed in the atmosphere, measurements made at such sites provide an integrated picture of large parts of the Earth including continents and city point sources. Note that this also applies to the other LLGHGs reported in Section 2.3 .
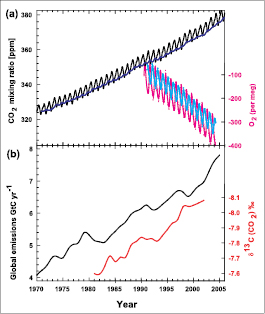
Figure 2.3. Recent CO2 concentrations and emissions. (a) CO2 concentrations (monthly averages) measured by continuous analysers over the period 1970 to 2005 from Mauna Loa, Hawaii (19°N, black; Keeling and Whorf, 2005 [NPR, SRC] ) and Baring Head, New Zealand (41°S, blue; following techniques by Manning et al., 1997 [NPR, ARC] ). Due to the larger amount of terrestrial biosphere in the NH, seasonal cycles in CO2 are larger there than in the SH. In the lower right of the panel, atmospheric oxygen (O2)measurements from flask samples are shown from Alert, Canada (82°N, pink) and Cape Grim, Australia (41°S, cyan) ( Manning and Keeling, 2006 [JoC, SRC] ). The O2 concentration is measured as ‘per meg’ deviations in the O2/N2 ratio from an arbitrary reference, analogous to the ‘per mil’ unit typically used in stable isotope work, but where the ratio is multiplied by 106 instead of 103 because much smaller changes are measured. (b) Annual global CO2 emissions from fossil fuel burning and cement manufacture in GtC yr–1 (black) through 2005, using data from the CDIAC website ( Marland et al, 2006 [NPR] ) to 2003 . Emissions data for 2004 and 2005 are extrapolated from CDIAC using data from the BP Statistical Review of World Energy ( BP, 2006 [NPR] ). Land use emissions are not shown; these are estimated to be between 0.5 and 2.7 GtC yr–1 for the 1990 s( Table 7.2 ). Annual averages of the 13C/12Cratio measured in atmospheric CO2 at Mauna Loa from 1981 to 2002 (red) are also shown ( Keeling et al, 2005 [NPR, SRC] ). The isotope data are expressed as δ13C(CO2)‰ (per mil) deviation from a calibration standard. Note that this scale is inverted to improve clarity.
In the 1980 s and 1990 s, it was recognised that greater coverage of CO2 measurements over continental areas was required to provide the basis for estimating sources and sinks of atmospheric CO2 over land as well as ocean regions. Because continuous CO2 analysers are relatively expensive to maintain and require meticulous on-site calibration, these records are now widely supplemented by air sample flask programmes, where air is collected in glass and metal containers at a large number of continental and marine sites. After collection, the filled flasks are sent to central well-calibrated laboratories for analysis. The most extensive network of international air sampling sites is operated by the National Oceanic and Atmospheric Administration’s Global Monitoring Division (NOAA/GMD; formerly NOAA/Climate Monitoring and Diagnostics Laboratory (CMDL)) in the USA. This organisation collates measurements of atmospheric CO2 from six continuous analyser locations as well as weekly flask air samples from a global network of almost 50 surface sites. Many international laboratories make atmospheric CO2 observations and worldwide databases of their measurements are maintained by the Carbon Dioxide Information Analysis Center (CDIAC) and by the World Data Centre for Greenhouse Gases (WDCGG) in the WMO Global Atmosphere Watch (GAW) programme. [6]
The increases in global atmospheric CO2 since the industrial revolution are mainly due to CO2 emissions from the combustion of fossil fuels, gas flaring and cement production. Other sources include emissions due to land use changes such as deforestation ( Houghton, 2003 [JoC, MoS] ) and biomass burning ( Andreae and Merlet, 2001 [JoC] ; van der Werf, 2004 [JoC] ). After entering the atmosphere, CO2 exchanges rapidly with the short-lived components of the terrestrial biosphere and surface ocean, and is then redistributed on time scales of hundreds of years among all active carbon reservoirs including the long-lived terrestrial biosphere and deep ocean. The processes governing the movement of carbon between the active carbon reservoirs, climate carbon cycle feedbacks and their importance in determining the levels of CO2 remaining in the atmosphere, are presented in Section 7.3 ,where carbon cycle budgets are discussed.
The increase in CO2 mixing ratios continues to yield the largest sustained RF of any forcing agent. The RF of CO2 is a function of the change in CO2 in the atmosphere over the time period under consideration. Hence, a key question is ‘How is the CO2 released from fossil fuel combustion, cement production and land cover change distributed amongst the atmosphere, oceans and terrestrial biosphere?’. This partitioning has been investigated using a variety of techniques. Among the most powerful of these are measurements of the carbon isotopes in CO2 as well as high-precision measurements of atmospheric oxygen (O2)content. The carbon contained in CO2 has two naturally occurring stable isotopes denoted 12Cand 13C. The first of these, 12C, is the most abundant isotope at about 99%, followed by 13Cat about 1%. Emissions of CO2 from coal, gas and oil combustion and land clearing have 13C/12Cisotopic ratios that are less than those in atmospheric CO2,and each carries a signature related to its source. Thus, as shown in Prentice et al. 2001 [NPR] ), when CO2 from fossil fuel combustion enters the atmosphere, the 13C/12Cisotopic ratio in atmospheric CO2 decreases at a predictable rate consistent with emissions of CO2 from fossil origin. Note that changes in the 13C/12Cratio of atmospheric CO2 are also caused by other sources and sinks, but the changing isotopic signal due to CO2 from fossil fuel combustion can be resolved from the other components ( Francey et al., 1995 [JoC] ). These changes can easily be measured using modern isotope ratio mass spectrometry, which has the capability of measuring 13C/12Cin atmospheric CO2 to better than 1 part in 105 ( Ferretti et al., 2000 [JoC, SRC] ). Data presented in Figure 2.3 for the 13C/12Cratio of atmospheric CO2 at Mauna Loa show a decreasing ratio, consistent with trends in both fossil fuel CO2 emissions and atmospheric CO2 mixing ratios ( Andres et al., 2000 [NPR, MoS] ; Keeling et al., 2005 [NPR, SRC] ).
AtmosphericO2 measurements provide a powerful and independent method of determining the partitioning of CO2 between the oceans and land ( Keeling et al., 1996 [JoC, SRC] ). AtmosphericO2 and CO2 changes are inversely coupled during plant respiration and photosynthesis. In addition, during the process of combustionO2 is removed from the atmosphere, producing a signal that decreases as atmospheric CO2 increases on a molar basis( Figure 2.3 ). Measuring changes in atmosphericO2 is technically challenging because of the difficulty of resolving changes at the part-per-million level in a background mixing ratio of roughly 209,000 ppm. These difficulties were first overcome by Keeling and Shertz 1992 [JoC, SRC] ), who used an interferometric technique to show that it is possible to track both seasonal cycles and the decline ofO2 in the atmosphere at the part-per-million level( Figure 2.3 ). Recent work by Manning and Keeling 2006 [JoC, SRC] ) indicates that atmosphericO2 is decreasing at a faster rate than CO2 is increasing, which demonstrates the importance of the oceanic carbon sink. Measurements of both the 13C/12Cratio in atmospheric CO2 and atmosphericO2 levels are valuable tools used to determine the distribution of fossil-fuel derived CO2 among the active carbon reservoirs, as discussed in Section 7.3 .In Figure 2.3 ,recent measurements in both hemispheres are shown to emphasize the strong linkages between atmospheric CO2 increases,O2 decreases, fossil fuel consumption and the 13C/12Cratio of atmospheric CO2.
From 1990 to 1999, a period reported in Prentice et al. 2001 [NPR] ), the emission rate due to fossil fuel burning and cement production increased irregularly from 6.1 to 6.5 GtC yr–1 or about 0.7% yr–1.From 1999 to 2005 however, the emission rate rose systematically from 6.5 to 7.8 GtC yr–1 ( BP, 2006 [NPR] ; Marland et al., 2006 [NPR] ) or about 3.0% yr–1,representing a period of higher emissions and growth in emissions than those considered in the TAR (see Figure 2.3 ). Carbon dioxide emissions due to global annual fossil fuel combustion and cement manufacture combined have increased by 70% over the last 30 years ( Marland et al., 2006 [NPR] ). The relationship between increases in atmospheric CO2 mixing ratios and emissions has been tracked using a scaling factor known as the apparent ‘airborne fraction’, defined as the ratio of the annual increase in atmospheric CO2 to the CO2 emissions from annual fossil fuel and cement manufacture combined ( Keeling et al., 1995 [JoC, SRC] ). On decadal scales, this fraction has averaged about 60% since the 1950 s. Assuming emissions of 7 GtC yr–1 and an airborne fraction remaining at about 60%, Hansen and Sato 2004 [JoC, ARC] ) predicted that the underlying long-term global atmospheric CO2 growth rate will be about 1.9 ppm yr–1,a value consistent with observations over the 1995 to 2005 decade.
Carbon dioxide emissions due to land use changes during the 1990 s are estimated as 0.5 to 2.7 GtC yr–1 ( Section 7.3 , Table 7.2 ), contributing 6% to 39% of the CO2 growth rate ( Brovkin et al., 2004 [ARC] Prentice et al. 2001 [NPR] ) cited an inventory-based estimate that land use change resulted in net emissions of 121 GtC between 1850 and 1990, after Houghton 1999 [JoC] , 2000 ). The estimate for this period was revised upwards to 134 GtC by Houghton 2003 [JoC, MoS] ), mostly due to an increase in estimated emissions prior to 1960 Houghton 2003 [JoC, MoS] ) also extended the inventory emissions estimate to 2000, giving cumulative emissions of 156 GtC since 1850 . In carbon cycle simulations by Brovkin et al. 2004 [ARC] and Matthews et al. 2004 [JoC, ARC] ), land use change emissions contributed 12 to 35 ppm of the total CO2 rise from 1850 to 2000 ( Section 2.5.3 , Table 2.8 ). Historical changes in land cover are discussed in Section 2.5.2 ,and the CO2 budget over the 1980 s and 1990 s is discussed further in Section 7.3 .
In 2005, the global mean average CO2 mixing ratio for the SIO network of 9 sites was 378.75 ± 0.13 ppm and for the NOAA/GMD network of 40 sites was 378.76 ± 0.05 ppm, yielding a global average of almost 379 ppm. For both networks, only sites in the remote marine boundary layer are used and high-altitude locations are not included. For example, the Mauna Loa site is excluded due to an ‘altitude effect’ of about 0.5 ppm. In addition, the 2005 values are still pending final reference gas calibrations used to measure the samples.
New measurements of CO2 from Antarctic ice and firn ( MacFarling Meure et al., 2006 [JoC] ) update and extend those from Etheridge et al. 1996 [JoC, SRC] ) to AD 0. The CO2 mixing ratio in 1750 was 277 ± 1.2 ppm. [7] This record shows variations between 272 and 284 ppm before 1800 and also that CO2 mixing ratios dropped by 5 to 10 ppm between 1600 and 1800 (see Section 6.3 ). The RF calculations usually take 1750 as the pre-industrial index (e.g., the TAR and this report). Therefore, using 1750 may slightly overestimate the RF, as the changes in the mixing ratios of CO2,CH4 andN2Oafter the end of this naturally cooler period may not be solely attributable to anthropogenic emissions. Using 1860 as an alternative start date for the RF calculations would reduce the LLGHG RF by roughly 10%. For the RF calculation, the data from Law Dome ice cap in the Antarctic are used because they show the highest age resolution (approximately 10 years) of any ice core record in existence. In addition, the high-precision data from the cores are connected to direct observational records of atmospheric CO2 from Cape Grim, Tasmania.
The simple formulae for RF of the LLGHG quoted in Ramaswamy et al. 2001 [NPR, MoS, SRC] ) are still valid. These formulae are based on global RF calculations where clouds, stratospheric adjustment and solar absorption are included, and give an RF of +3.7 Wm–2 for a doubling in the CO2 mixing ratio. (The formula used for the CO2 RF calculation in this chapter is the IPCC 1990 [NPR] ) expression as revised in the TAR. Note that for CO2,RF increases logarithmically with mixing ratio.) Collins et al. 2006 [JoC, MoS, SRC] ) performed a comparison of five detailed line-by-line models and 20 GCM radiation schemes. The spread of line-by-line model results were consistent with the ±10% uncertainty estimate for the LLGHG RFs adopted in Ramaswamy et al. 2001 [NPR, MoS, SRC] ) and a similar ±10% for the 90% confidence interval is adopted here. However, it is also important to note that these relatively small uncertainties are not always achievable when incorporating the LLGHG forcings into GCMs. For example, both Collins et al. 2006 [JoC, MoS, SRC] and Forster and Taylor 2006 [JoC, MoS, SRC] ) found that GCM radiation schemes could have inaccuracies of around 20% in their total LLGHG RF (see also Sections 2.3.2 and 10.2 ).
Using the global average value of 379 ppm for atmospheric CO2 in 2005 gives an RF of 1.66 ± 0.17 Wm–2;a contribution that dominates that of all other forcing agents considered in this chapter. This is an increase of 13 to 14% over the value reported for 1998 in Ramaswamy et al. 2001 [NPR, MoS, SRC] ). This change is solely due to increases in atmospheric CO2 and is also much larger than the RF changes due to other agents. In the decade 1995 to 2005, the RF due to CO2 increased by about 0.28 Wm–2 (20%), an increase greater than that calculated for any decade since at least 1800 (see Section 6.6 and FAQ 2.1 ,Figure 1).
Table 2.1 summarises the present-day mixing ratios and RF for the LLGHGs, and indicates changes since 1998 . The RF from CO2 and that from the other LLGHGs have a high level of scientific understanding( Section 2.9 , Table 2.11 ). Note that the uncertainty in RF is almost entirely due to radiative transfer assumptions and not mixing ratio estimates, therefore trends in RF can be more accurately determined than the absolute RF. From Section 2.5.3 , Table 2.8 ,the contribution from land use change to the present CO2 RF is likely to be about 0.4 Wm–2 (since 1850 ). This implies that fossil fuel and cement production have likely contributed about three-quarters of the current RF.
Table 2.1 .Present-day concentrations and RF for the measured LLGHGs. The changes since 1998 (the time of the TAR estimates) are also shown.
| Concentrationsb and their changesc | Radiative Forcingd | |||
|---|---|---|---|---|
| Speciesa | 2005 | Change since 1998 | 2005 (Wm–2) | Change since1998 (%) |
| CO2 | 379 ± 0.65 ppm | +13 ppm | 1.66 | +13 |
| CH4 | 1,774 ± 1.8 ppb | +11 ppb | 0.48 | - |
| N2O | 319 ± 0.12 ppb | +5 ppb | 0.16 | +11 |
| ppt | ppt | |||
| CFC-11 | 251 ± 0.36 | –13 | 0.063 | –5 |
| CFC-12 | 538 ± 0.18 | +4 | 0.17 | +1 |
| CFC-113 | 79 ± 0.064 | –4 | 0.024 | –5 |
| HCFC-22 | 169 ± 1.0 | +38 | 0.033 | +29 |
| HCFC-141b | 18 ± 0.068 | +9 | 0.0025 | +93 |
| HCFC-142b | 15 ± 0.13 | +6 | 0.0031 | +57 |
| CH3CCl3 | 19 ± 0.47 | –47 | 0.0011 | –72 |
| CCl4 | 93 ± 0.17 | –7 | 0.012 | –7 |
| HFC-125 | 3.7 ± 0.10e | +2.6f | 0.0009 | +234 |
| HFC-134a | 35 ± 0.73 | +27 | 0.0055 | +349 |
| HFC-152a | 3.9 ± 0.11e | +2.4f | 0.0004 | +151 |
| HFC-23 | 18 ± 0.12g,h | +4 | 0.0033 | +29 |
| SF6 | 5.6 ± 0.038i | +1.5 | 0.0029 | +36 |
| CF4 (PFC-14) | 74 ± 1.6j | - | 0.0034 | - |
| C2F6 (PFC-116) | 2.9 ± 0.025g,h | +0.5 | 0.0008 | +22 |
| CFCs Totalk | 0.268 | –1 | ||
| HCFCs Total | 0.039 | +33 | ||
| Montreal Gases | 0.320 | –1 | ||
| Other Kyoto Gases(HFCs + PFCs + SF6) | 0.017 | +69 | ||
| Halocarbons | 0.337 | +1 | ||
| Total LLGHGs | 2.63 | +9 | ||
2.3.2 Atmospheric Methane
This section describes the current global measurement programmes for atmospheric CH4,which provide the data required for the understanding of its budget and for the calculation of its RF. In addition, this section provides data for the pre-industrial levels of CH4 required as the accepted reference level for these calculations. Detailed analyses of CH4 budgets and its biogeochemistry are presented in Section 7.4 .
Methane has the second-largest RF of the LLGHGs after CO2 ( Ramaswamy et al., 2001 [NPR, MoS, SRC] ). Over the last 650 kyr, ice core records indicate that the abundance of CH4 in the Earth’s atmosphere has varied from lows of about 400 ppb during glacial periods to highs of about 700 ppb during interglacials ( Spahni et al., 2005 [JoC, ARC] ) with a single measurement from the Vostok core reaching about 770 ppb (see Figure 6.3 ).
In 2005, the global average abundance of CH4 measured at the network of 40 surface air flask sampling sites operated by NOAA/GMD in both hemispheres was 1,774.62 ± 1.22 ppb. [8] This is the most geographically extensive network of sites operated by any laboratory and it is important to note that the calibration scale it uses has changed since the TAR ( Dlugokencky et al., 2005 [JoC, SRC] ). The new scale (known as NOAA04) increases all previously reported CH4 mixing ratios from NOAA/GMD by about 1%, bringing them into much closer agreement with the Advanced Global Atmospheric Gases Experiment (AGAGE) network. This scale will be used by laboratories participating in the WMO’s GAW programme as a ‘common reference’. Atmospheric CH4 is also monitored at five sites in the NH and SH by the AGAGE network. This group uses automated systems to make 36 CH4 measurements per day at each site, and the mean for 2005 was 1,774.03 ± 1.68 ppb with calibration and methods described by Cunnold et al. 2002 [JoC] ). For the NOAA/GMD network, the 90% confidence interval is calculated with a Monte Carlo technique, which only accounts for the uncertainty due to the distribution of sampling sites. For both networks, only sites in the remote marine boundary layer are used and continental sites are not included. Global databases of atmospheric CH4 measurements for these and other CH4 measurement programmes (e.g., Japanese, European and Australian) are maintained by the CDIAC and by the WDCGG in the GAW programme.
Present atmospheric levels of CH4 are unprecedented in at least the last 650 kyr ( Spahni et al., 2005 [JoC, ARC] ). Direct atmospheric measurements of the gas made at a wide variety of sites in both hemispheres over the last 25 years show that, although the abundance of CH4 has increased by about 30% during that time, its growth rate has decreased substantially from highs of greater than 1% yr–1 in the late 1 70s and early 1980 s ( Blake and Rowland, 1988 [JoC] ) to lows of close to zero towards the end of the 1990 s ( Dlugokencky et al., 1998 [JoC, SRC] ; Simpson et al., 2002 [JoC] ). The slowdown in the growth rate began in the 1980 s, decreasing from 14 ppb yr–1 (about 1% yr–1)in 1984 to close to zero during 1999 to 2005, for the network of surface sites maintained by NOAA/GMD ( Dlugokencky et al., 2003 [JoC, SRC] ). Measurements by Lowe et al. 2004 [JoC, SRC] ) for sites in the SH and Cunnold et al. 2002 [JoC] ) for the network of GAGE/AGAGE sites show similar features. A key feature of the global growth rate of CH4 is its current interannual variability, with growth rates ranging from a high of 14 ppb yr–1 in 1998 to less than zero in 2001, 2004 and 2005 .( Figure 2.4 )
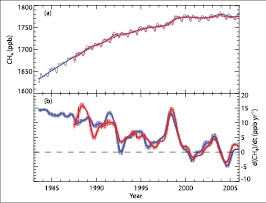
Figure 2.4. Recent CH4 concentrations and trends. (a) Time series of global CH4 abundance mole fraction (in ppb) derived from surface sites operated by NOAA/GMD (blue lines) and AGAGE (red lines). The thinner lines show the CH4 global averages and the thicker lines are the de-seasonalized global average trends from both networks. (b) Annual growth rate (ppb yr–1)in global atmospheric CH4 abundance from 1984 through the end of 2005 (NOAA/GMD, blue), and from 1988 to the end of 2005 (AGAGE, red). To derive the growth rates and their uncertainties for each month, a linear least squares method that takes account of the autocorrelation of residuals is used. This follows the methods of Wang et al. 2002 [JoC] ) and is applied to the de-seasonalized global mean mole fractions from (a) for values six months before and after the current month. The vertical lines indicate ±2 standard deviation uncertainties (95% confidence interval), and 1 standard deviation uncertainties are between 0.1 and 1.4 ppb yr–1 for both AGAGE and NOAA/GMD. Note that the differences between AGAGE and NOAA/GMD calibration scales are determined through occasional intercomparisons.
The reasons for the decrease in the atmospheric CH4 growth rate and the implications for future changes in its atmospheric burden are not understood ( Prather et al., 2001 [NPR, ARC] ) but are clearly related to changes in the imbalance between CH4 sources and sinks. Most CH4 is removed from the atmosphere by reaction with the hydroxyl free radical (OH), which is produced photochemically in the atmosphere. The role of OH in controlling atmospheric CH4 levels is discussed in Section 2.3.5 .Other minor sinks include reaction with free chlorine ( Platt et al., 2004 [SRC] ; Allan et al., 2005 [JoC] ), destruction in the stratosphere and soil sinks ( Born et al., 1990 [JoC] ).
The total global CH4 source is relatively well known but the strength of each source component and their trends are not. As detailed in Section 7.4 ,the sources are mostly biogenic and include wetlands, rice agriculture, biomass burning and ruminant animals. Methane is also emitted by various industrial sources including fossil fuel mining and distribution. Prather et al. 2001 [NPR, ARC] ) documented a large range in ‘bottom-up’ estimates for the global source of CH4.New source estimates published since then are documented in Table 7.6 .However, as reported by Bergamaschi et al. 2005 [MoS] ), national inventories based on ‘bottom-up’ studies can grossly underestimate emissions and ‘top-down’ measurement-based assessments of reported emissions will be required for verification. Keppler et al. 2006 [JoC] ) reported the discovery of emissions of CH4 from living vegetation and estimated that this contributed 10 to 30% of the global CH4 source. This work extrapolates limited measurements to a global source and has not yet been confirmed by other laboratories, but lends some support to space-borne observations of CH4 plumes above tropical rainforests reported by Frankenberg et al. 2005 [JoC] ). That such a potentially large source of CH4 could have been missed highlights the large uncertainties involved in current ‘bottom-up’ estimates of components of the global source (see Section 7.4 ).
Several wide-ranging hypotheses have been put forward to explain the reduction in the CH4 growth rate and its variability. For example, Hansen et al. 2000 [JoC, ARC] ) considered that economic incentives have led to a reduction in anthropogenic CH4 emissions. The negligible long-term change in its main sink (OH; see Section 2.3.5 and Figure 2.8 )implies that CH4 emissions are not increasing. Similarly, Dlugokencky et al. 1998 [JoC, SRC] and Francey et al. 1999 [JoC] ) suggested that the slowdown in the growth rate reflects a stabilisation of CH4 emissions, given that the observations are consistent with stable emissions and lifetime since 1982 .
Relatively large anomalies occurred in the growth rate during 1991 and 1998, with peak values reaching 15 and 14 ppb yr–1,respectively (about 1% yr–1). The anomaly in 1991 was followed by a dramatic drop in the growth rate in 1992 and has been linked with the Mt. Pinatubo volcanic eruption in June 1991, which injected large amounts of ash and (sulphur dioxide) SO2 into the lower stratosphere of the tropics with subsequent impacts on tropical photochemistry and the removal of CH4 by atmospheric OH ( Bekki et al., 1994 [JoC] ; Dlugokencky et al., 1996 [JoC, SRC] Lelieveld et al. 1998 [JoC, MoS, ARC] and Walter et al. 2001a [JoC, MoS, ARC] ,b) proposed that lower temperatures and lower precipitation in the aftermath of the Mt. Pinatubo eruption could have suppressed CH4 emissions from wetlands. At this time, and in parallel with the growth rate anomaly in the CH4 mixing ratio, an anomaly was observed in the 13C/12Cratio of CH4 at surface sites in the SH. This was attributed to a decrease in emissions from an isotopically heavy source such as biomass burning ( Lowe et al., 1997 [JoC, SRC] ; Mak et al., 2000 [JoC, SRC] ), although these data were not confirmed by lower frequency measurements from the same period made by Francey et al. 1999 [JoC] ).
For the relatively large increase in the CH4 growth rate reported for 1998, Dlugokencky et al. 2001 [JoC, SRC] ) suggested that wetland and boreal biomass burning sources might have contributed to the anomaly, noting that 1998 was the warmest year globally since surface instrumental temperature records began. Using an inverse method, Chen and Prinn 2006 [JoC, MoS, SRC] ) attributed the same event primarily to increased wetland and rice region emissions and secondarily to biomass burning. The same conclusion was reached by Morimoto et al. 2006 [JoC] ), who used carbon isotopic measurements of CH4 to constrain the relative contributions of biomass burning (one-third) and wetlands (two-thirds) to the increase.
Based on ice core measurements of CH4 ( Etheridge et al., 1998 [JoC, SRC] ), the pre-industrial global value for CH4 from 1700 to 1800 was 715 ± 4 ppb (it was also 715 ± 4 ppb in 1750 ), thus providing the reference level for the RF calculation. This takes into account the inter-polar difference in CH4 as measured from Greenland and Antarctic ice cores.
The RF due to changes in CH4 mixing ratio is calculated with the simplified yet still valid expression for CH4 given in Ramaswamy et al. 2001 [NPR, MoS, SRC] ). The change in the CH4 mixing ratio from 715 ppb in 1750 to 1,774 ppb (the average mixing ratio from the AGAGE and GMD networks) in 2005 gives an RF of +0.48 ± 0.05 Wm–2,ranking CH4 as the second highest RF of the LLGHGs after CO2 ( Table 2.1 ). The uncertainty range in mixing ratios for the present day represents intra-annual variability, which is not included in the pre-industrial uncertainty estimate derived solely from ice core sampling precision. The estimate for the RF due to CH4 is the same as in Ramaswamy et al. 2001 [NPR, MoS, SRC] ) despite the small increase in its mixing ratio. The spectral absorption by CH4 is overlapped to some extent byN2Olines (taken into account in the simplified expression). Taking the overlapping lines into account using currentN2Omixing ratios instead of pre-industrial mixing ratios (as in Ramaswamy et al., 2001 [NPR, MoS, SRC] ) reduces the current RF due to CH4 by 1%.
Collins et al. 2006 [JoC, MoS, SRC] ) confirmed that line-by-line models agree extremely well for the calculation of clear-sky instantaneous RF from CH4 andN2Owhen the same atmospheric background profile is used. However, GCM radiation schemes were found to be in poor agreement with the line-by-line models, and errors of over 50% were possible for CH4,N2Oand the CFCs. In addition, a small effect from the absorption of solar radiation was found with the line-by-line models, which the GCMs did not include( Section 10.2 ).
2.3.3 Other Kyoto Protocol Gases
At the time of the TAR,N2Ohad the fourth largest RF among the LLGHGs behind CO2,CH4 and CFC-12. The TAR quoted an atmosphericN2Oabundance of 314 ppb in 1998, an increase of 44 ppb from its pre-industrial level of around 270 ± 7 ppb, which gave an RF of +0.15 ± 0.02 Wm–2.This RF is affected by atmospheric CH4 levels due to overlapping absorptions. AsN2Ois also the major source of ozone-depleting nitric oxide (NO) and nitrogen dioxide (NO2)in the stratosphere, it is routinely reviewed in the ozone assessments; the most recent assessment ( Montzka et al., 2003 [NPR, SRC] ) recommended an atmospheric lifetime of 114 years forN2O. The TAR pointed out large uncertainties in the major soil, agricultural, combustion and oceanic sources ofN2O. Given these emission uncertainties, its observed rate of increase of 0.2 to 0.3% yr–1 was not inconsistent with its better-quantified major sinks (principally stratospheric destruction). The primary driver for the industrial era increase ofN2Owas concluded to be enhanced microbial production in expanding and fertilized agricultural lands.
Ice core data forN2Ohave been reported extending back 2,000 years and more before present ( MacFarling Meure et al., 2006 [JoC] ; Section 6.6 ). These data, as for CO2 and CH4,show relatively little changes in mixing ratios over the first 1,800 years of this record, and then exhibit a relatively rapid rise (see FAQ 2.1 ,Figure 1). Since 1998, atmosphericN2Olevels have steadily risen to 319 ± 0.12 ppb in 2005, and levels have been increasing approximately linearly (at around 0.26% yr–1)for the past few decades( Figure 2.5 ). A change in theN2Omixing ratio from 270 ppb in 1750 to 319 ppb in 2005 results in an RF of +0.16 ± 0.02 Wm–2,calculated using the simplified expression given in Ramaswamy et al. 2001 [NPR, MoS, SRC] ). The RF has increased by 11% since the time of the TAR( Table 2.1 ). As CFC-12 levels slowly decline (see Section 2.3.4 ),N2Oshould, with its current trend, take over third place in the LLGHG RF ranking.
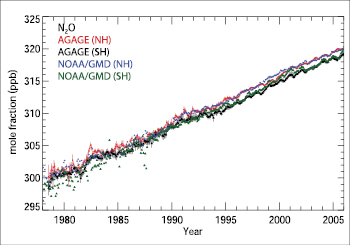
Figure 2.5. Hemispheric monthly mean N2Omole fractions (ppb) (crosses for the NH and triangles for the SH). Observations (in situ) of N2Ofrom the Atmospheric Lifetime Experiment (ALE) and GAGE (through the mid- 1990 s) and AGAGE (since the mid- 1990 s) networks ( Prinn et al., 2000 [JoC, SRC] , 2005b ) are shown with monthly standard deviations. Data from NOAA/GMD are shown without these standard deviations ( Thompson et al., 2004 [NPR] ). The general decrease in the variability of the measurements over time is due mainly to improved instrumental precision. The real signal emerges only in the last decade.
Since the TAR, understanding of regionalN2Ofluxes has improved. The results of various studies that quantified the globalN2Oemissions from coastal upwelling areas, continental shelves, estuaries and rivers suggest that these coastal areas contribute 0.3 to 6.6 TgN yr–1 ofN2Oor 7 to 61% of the total oceanic emissions ( Bange et al., 1996 [JoC] ; Nevison et al., 2004b [JoC, SRC] ; Kroeze et al., 2005 [MoS] ; see also Section 7.4 ). Using inverse methods and AGAGE Ireland measurements, Manning et al. 2003 [JoC, MoS] ) estimated EUN2Oemissions of 0.9 ± 0.1 TgN yr–1 that agree well with the United Nations Framework Convention on Climate Change (UNFCCC)N2Oinventory (0.8 ± 0.1 TgN yr–1 Melillo et al. 2001 [JoC] ) provided evidence from Brazilian land use sequences that the conversion of tropical forest to pasture leads to an initial increase but a later decline in emissions ofN2Orelative to the original forest. They also deduced that Brazilian forest soils alone contribute about 10% of total globalN2Oproduction. Estimates ofN2Osources and sinks using observations and inverse methods had earlier implied that a large fraction of globalN2Oemissions in 1978 to 1988 were tropical: specifically 20 to 29% from 0° to 30°S and 32 to 39% from 0° to 30°N compared to 11 to 15% from 30°S to 90°S and 22 to 34% from 30°N to 90°N ( Prinn et al., 1990 [JoC, SRC] ). These estimates were uncertain due to their significant sensitivity to assumed troposphere-stratosphere exchange rates that strongly influence inter-hemispheric gradients. Hirsch et al. 2006 [JoC, MoS] ) used inverse modelling to estimate significantly lower emissions from 30°S to 90°S (0 to 4%) and higher emissions from 0° to 30°N (50 to 64%) than Prinn et al. 1990 [JoC, SRC] ) during 1998 to 2001, with 26 to 36% from the oceans. The stratosphere is also proposed to play an important role in the seasonal cycles ofN2O(Nevison et al., 2004a ). For example, its well-defined seasonal cycle in the SH has been interpreted as resulting from the net effect of seasonal oceanic outgassing of microbially producedN2O, stratospheric intrusion of low-N2Oair and other processes ( Nevison et al., 2005 [JoC] ). Nevison et al. also estimated a Southern Ocean (30°S–90°S)N2Osource of 0.9 TgN yr–1,or about 5% of the global total. The complex seasonal cycle in the NH is more difficult to reconcile with seasonal variations in the northern latitude soil sources and stratospheric intrusions ( Prinn et al., 2000 [JoC, SRC] ; T. Liao et al., 2004 [Ambiguous] ). The destruction ofN2Oin the stratosphere causes enrichment of its heavier isotopomers and isotopologues, providing a potential method to differentiate stratospheric and surface flux influences on troposphericN2O(Morgan et al., 2004 ).
Human-made PFCs, HFCs and SF6 are very effective absorbers of infrared radiation, so that even small amounts of these gases contribute significantly to the RF of the climate system. The observations and global cycles of the major HFCs, PFCs and SF6 were reviewed in Velders et al. 2005 [NPR] ), and this section only provides a brief review and an update for these species. Table 2.1 shows the present mixing ratio and recent trends in the halocarbons and their RFs. Absorption spectra of most halocarbons reviewed here and in the following section are characterised by strongly overlapping spectral lines that are not resolved at tropospheric pressures and temperatures, and there is some uncertainty in cross section measurements. Apart from the uncertainties stemming from the cross sections themselves, differences in the radiative flux calculations can arise from the spectral resolution used, tropopause heights, vertical, spatial and seasonal distributions of the gases, cloud cover and how stratospheric temperature adjustments are performed. IPCC/TEAP ( 2005 ) concluded that the discrepancy in the RF calculation for different halocarbons, associated with uncertainties in the radiative transfer calculation and the cross sections, can reach 40%. Studies reviewed in IPCC/TEAP ( 2005 ) for the more abundant HFCs show that an agreement better than 12% can be reached for these when the calculation conditions are better constrained (see Section 2.10.2 ).
The HFCs of industrial importance have lifetimes in the range 1.4 to 270 years. The HFCs with the largest observed mole fractions in 1998 (as reported in the TAR) were, in descending order, HFC-23, HFC-134a and HFC-152a. In 2005, the observed mixing ratios of the major HFCs in the atmosphere were 35 ppt for HFC-134a, 17.5 ppt for HFC-23 ( 2003 value), 3.7 ppt for HFC-125 and 3.9 ppt for HFC-152a( Table 2.1 ). Within the uncertainties in calibration and emissions estimates, the observed mixing ratios of the HFCs in the atmosphere can be explained by the anthropogenic emissions. Measurements are available from GMD ( Thompson et al., 2004 [NPR] ) and AGAGE ( Prinn et al., 2000 [JoC, SRC] ; O’Doherty et al., 2004 [JoC] ; Prinn et al., 2005b [NPR, SRC] ) networks as well as from University of East Anglia (UEA) studies in Tasmania (updated from Oram et al., 1998 [JoC, SRC] ; Oram, 1999 [NPR, MoS, SRC] ). These data, summarised in Figure 2.6 ,show a continuation of positive HFC trends and increasing latitudinal gradients (larger trends in the NH) due to their predominantly NH sources. The air conditioning refrigerant HFC-134a is increasing at a rapid rate in response to growing emissions arising from its role as a replacement for some CFC refrigerants. With a lifetime of about 14 years, its current trends are determined primarily by its emissions and secondarily by its atmospheric destruction. Emissions of HFC-134a estimated from atmospheric measurements are in approximate agreement with industry estimates ( Huang and Prinn, 2002 [JoC, MoS, SRC] ; O’Doherty et al., 2004 [JoC] ). IPCC/TEAP ( 2005 ) reported that global HFC-134a emissions started rapidly increasing in the early 1990 s, and that in Europe, sharp increases in emissions are noted for HFC-134a from 1995 to 1998 and for HFC-152a from 1996 to 2000, with some levelling off through 2003 . The concentration of the foam blower HFC-152a, with a lifetime of only about 1.5 years, is rising approximately exponentially, with the effects of increasing emissions only partly offset by its rapid atmospheric destruction. Hydrofluorocarbon-23 has a very long atmospheric lifetime (approximately 270 years) and is mainly produced as a by-product of HCFC-22 production. Its concentrations are rising approximately linearly, driven by these emissions, with its destruction being only a minor factor in its budget. There are also smaller but rising concentrations of HFC-125 and HFC-143a, which are both refrigerants.
The PFCs, mainly CF4 (PFC-14) andC2F6 (PFC-116), and SF6 have very large radiative efficiencies and lifetimes in the range 1,000 to 50,000 years (see Section 2.10 , Table 2.14 ), and make an essentially permanent contribution to RF. The SF6 andC2F6 concentrations and RFs have increased by over 20% since the TAR( Table 2.1 and Figure 2.6 ), but CF4 concentrations have not been updated since 1997 . Both anthropogenic and natural sources of CF4 are important to explain its observed atmospheric abundance. These PFCs are produced as by-products of traditional aluminium production, among other activities. The CF4 concentrations have been increasing linearly since about 1960 and CF4 has a natural source that accounts for about one-half of its current atmospheric content ( Harnisch et al., 1996 [JoC] ). Sulphur hexafluoride (SF6)is produced for use as an electrical insulating fluid in power distribution equipment and also deliberately released as an essentially inert tracer to study atmospheric and oceanic transport processes. Its concentration was 4.2 ppt in 1998 (TAR) and has continued to increase linearly over the past decade, implying that emissions are approximately constant. Its very long lifetime ensures that its emissions accumulate essentially unabated in the atmosphere.
2.3.4 Montreal Protocol Gases
The Montreal Protocol on Substances that Deplete the Ozone Layer regulates many radiatively powerful greenhouse gases for the primary purpose of lowering stratospheric chlorine and bromine concentrations. These gases include the CFCs, HCFCs, chlorocarbons, bromocarbons and halons. Observations and global cycles of these gases were reviewed in detail in Chapter1 of the 2002 Scientific Assessment of Ozone Depletion ( WMO, 2003 [NPR] ) and in IPCC/TEAP ( 2005 ). The discussion here focuses on developments since these reviews and on those gases that contribute most to RF rather than to halogen loading. Using observed 2005 concentrations, the Montreal Protocol gases have contributed 12% (0.320 Wm–2)to the direct RF of all LLGHGs and 95% to the halocarbon RF( Table 2.1 ). This contribution is dominated by the CFCs. The effect of the Montreal Protocol on these gases has been substantial. IPCC/TEAP ( 2005 ) concluded that the combined CO2-equivalent emissions of CFCs, HCFCs and HFCs decreased from a peak of about 7.5 GtCO2-eq yr–1 in the late 1980 s to about 2.5 GtCO2-eq yr–1 by the year 2000, corresponding to about 10% of that year’s CO2 emissions due to global fossil fuel burning.
Measurements of the CFCs and HCFCs, summarised in Figure 2.6 ,are available from the AGAGE network ( Prinn et al., 2000 [JoC, SRC] , 2005b ) and the GMD network ( Montzka et al., 1999 [JoC, MoS, SRC] updated; Thompson et al., 2004 [NPR] ). Certain flask measurements are also available from the University of California at Irvine (UCI; Blake et al., 2001 [JoC] updated) and UEA ( Oram et al., 1998 [JoC, SRC] ; Oram, 1999 [NPR, MoS, SRC] updated). Two of the major CFCs (CFC-11 and CFC-113) have both been decreasing in the atmosphere since the mid- 1990 s. While their emissions have decreased very substantially in response to the Montreal Protocol, their long lifetimes of around 45 and 85 years, respectively, mean that their sinks can reduce their levels by only about 2% and 1% yr–1,respectively. Nevertheless, the effect of the Montreal Protocol has been to substantially reduce the growth of the halocarbon RF, which increased rapidly from 1950 until about 1990 . The other major CFC (CFC-12), which is the third most important LLGHG, is finally reaching a plateau in its atmospheric levels (emissions equal loss) and may have peaked in 2003 . Its 100-year lifetime means that it can decrease by only about 1% yr–1 even when emissions are zero. The levelling off for CFC-12 and approximately linear downward trends for CFC-11 and CFC-113 continue. Latitudinal gradients of all three are very small and decreasing as expected. The combined CFC and HCFC RF has been slowly declining since 2003 . Note that the 1998 concentrations of CFC-11 and CFC-12 were overestimated in Table 6.1 of the TAR. This means that the total halocarbon RF quoted for 2005 in Table 2.1 (0.337 Wm–2)is slightly smaller than the 0.34 Wm–2 quoted in the TAR, even though the measurements indicate a small 1% rise in the total halocarbon RF since the time of the TAR( Table 2.1 ).
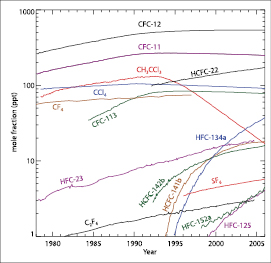
Figure 2.6. Temporal evolution of the global average dry-air mole fractions (ppt) of the major halogen-containing LLGHGs. These are derived mainly using monthly mean measurements from the AGAGE and NOAA/GMD networks. For clarity, the two network values are averaged with equal weight when both are available. While differences exist, these network measurements agree reasonably well with each other (except for CCl4 (differences of 2 – 4% between networks) and HCFC-142b (differences of 3 – 6% between networks)), and with other measurements where available (see text for references for each gas).
The major solvent, methyl chloroform (CH3CCl3), is of special importance regarding RFs, not because of its small RF (see Table 2.1 and Figure 2.6 ), but because this gas is widely used to estimate concentrations of OH, which is the major sink species for CH4,HFCs, and HCFCs and a major production mechanism for sulphate, nitrate and some organic aerosols as discussed in Section 2.3.5 .The global atmospheric methyl chloroform concentration rose steadily from 1978 to reach a maximum in 1992 ( Prinn et al., 2001 [JoC, SRC] ; Montzka et al., 2003 [NPR, SRC] ). Since then, concentrations have decreased rapidly, driven by a relatively short lifetime of 4.9 years and phase-out under the Montreal Protocol, to levels in 2003 less than 20% of the levels when AGAGE measurements peaked in 1992 ( Prinn et al., 2005a [JoC, SRC] ). Emissions of methyl chloroform determined from industry data ( (McCulloch and Midgley, 2001 ) ) may be too small in recent years. The 2000 to 2003 emissions from Europe estimated using surface observations ( Reimann et al., 2005 [JoC] ) show that 1.2 to 2.3 Gg yr–1 need to be added over this 4-year period to the above industry estimates for Europe. Estimates of European emissions in 2000 exceeding 20 Gg ( Krol et al., 2003 [JoC] ) are not supported by analyses of the above extensive surface data ( Reimann et al., 2005 [JoC] ). From multi-year measurements, Li et al. 2005 [JoC, MoS] ) estimated 2001 to 2002 emissions from the USA of 2.2 Gg yr–1 (or about half of those estimated from more temporally but less geographically limited measurements by Millet and Goldstein, 2004 [JoC] ), and suggested that 1996 to 1998 US emissions may be underestimated by an average of about 9.0 Gg yr–1 over this 3-year period. East Asian emissions deduced from aircraft data in 2001 are about 1.7 Gg above industry data ( Palmer et al., 2003 [JoC] ; see also Yokouchi et al., 2005 ) while recent Australian and Russian emissions are negligible ( Prinn et al., 2001 [JoC, SRC] ; Hurst et al., 2004 [JoC] ).
Carbon tetrachloride (CCl4)is the second most rapidly decreasing atmospheric chlorocarbon after methyl chloroform. Levels peaked in early 1990 and decreased approximately linearly since then( Figure 2.7 ). Its major use was as a feedstock for CFC manufacturing. Unlike methyl chloroform, a significant inter-hemispheric CCl4 gradient still exists in 2005 in spite of its moderately long lifetime of 20 to 30 years, resulting from a persistence of significant NH emissions.
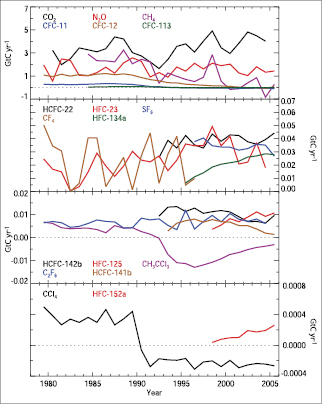
Figure 2.7. Annual rates of change in the global atmospheric masses of each of the major LLGHGs expressed in common units of GtC yr–1.These rates are computed from their actual annual mass changes in Gt yr–1 (as derived from their observed global and annual average mole fractions presented in Figures 2.3 to 2.6 and discussed in Sections 2.3.1 to 2.3.4 )by multiplying them by their GWPs for 100-year time horizons and then dividing by the ratio of the CO2 to carbon (C) masses (44/12). These rates are positive or negative whenever the mole fractions are increasing or decreasing, respectively. Use of these common units provides an approximate way to intercompare the fluxes of LLGHGs, using the same approach employed to intercompare the values of LLGHG emissions under the Kyoto Protocol (see, e.g., Prinn, 2004 [NPR, SRC] ). Note that the negative indirect RF of CFCs and HCFCs due to stratospheric ozone depletion is not included. The oscillations in the CF4 curve may result partly from truncation in reported mole fractions.
HCFCs of industrial importance have lifetimes in the range of 1.3 to 20 years. Global and regional emissions of the CFCs and HCFCs have been derived from observed concentrations and can be used to check emission inventory estimates. Montzka et al. 2003 [NPR, SRC] ) and IPCC/TEAP ( 2005 ) concluded that global emissions of HCFC-22 rose steadily over the period 1975 to 2000, while those of HCFC-141b and HCFC-142b started increasing quickly in the early 1990 s and then began to decrease after 2000 .
To provide a direct comparison of the effects on global warming due to the annual changes in each of the non-CO2 greenhouse gases (discussed in Sections 2.3.2 , 2.3.3 and 2.3.4 )relative to CO2, Figure 2.7 shows these annual changes in atmospheric mass multiplied by the GWP (100-year horizon) for each gas (e.g., Prinn, 2004 [NPR, SRC] ). By expressing them in this way, the observed changes in all non-CO2 gases in GtC equivalents and the significant roles of CH4,N2Oand many halocarbons are very evident. This highlights the importance of considering the full suite of greenhouse gases for RF calculations.
2.3.5 Trends in the Hydroxyl Free Radical
The hydroxyl free radical (OH) is the major oxidizing chemical in the atmosphere, destroying about 3.7 Gt of trace gases, including CH4 and all HFCs and HCFCs, each year ( Ehhalt, 1999 [NPR] ). It therefore has a very significant role in limiting the LLGHG RF. IPCC/TEAP ( 2005 ) concluded that the OH concentration might change in the 21st century by –18 to +5% depending on the emission scenario. The large-scale concentrations and long-term trends in OH can be inferred indirectly using global measurements of trace gases for which emissions are well known and the primary sink is OH. The best trace gas used to date for this purpose is methyl chloroform; long-term measurements of this gas are reviewed in Section 2.3.4 .Other gases that are useful OH indicators include 14CO, which is produced primarily by cosmic rays ( Lowe and Allan, 2002 [SRC] ). While the accuracy of the 14CO cosmic ray and other 14CO source estimates and the frequency and spatial coverage of its measurements do not match those for methyl chloroform, the 14CO lifetime (2 months) is much shorter than that of methyl chloroform (4.9 years). As a result, 14CO provides estimates of average concentrations of OH that are more regional, and is capable of resolving shorter time scales than those estimated from methyl chloroform. The 14CO source variability is better defined than its absolute magnitude so it is better for inferring relative rather than absolute trends. Another useful gas is the industrial chemical HCFC-22. It yields OH concentrations similar to those derived from methyl chloroform, but with less accuracy due to greater uncertainties in emissions and less extensive measurements ( Miller et al., 1998 [JoC] ). The industrial gases HFC-134a, HCFC-141b and HCFC-142b are potentially useful OH estimators, but the accuracy of their emission estimates needs improvement ( Huang and Prinn, 2002 [JoC, MoS, SRC] ; O’Doherty et al., 2004 [JoC] ).
Indirect measurements of OH using methyl chloroform have established that the globally weighted average OH concentration in the troposphere is roughly 106 radicals per cubic centimetre ( Prinn et al., 2001 [JoC, SRC] ; Krol and Lelieveld, 2003 [JoC] ). A similar average concentration is derived using 14CO ( Quay et al., 2000 [JoC] ), although the spatial weighting here is different. Note that methods to infer global or hemispheric average OH concentrations may be insensitive to compensating regional OH changes such as OH increases over continents and decreases over oceans ( Lelieveld et al., 2002 [JoC, ARC] ). In addition, the quoted absolute OH concentrations (but not their relative trends) depend on the choice of weighting (e.g., ( Lawrence et al., 2001 ) ). While the global average OH concentration appears fairly well defined by these indirect methods, the temporal trends in OH are more difficult to discern since they require long-term measurements, optimal inverse methods and very accurate calibrations, model transports and methyl chloroform emissions data. From AGAGE methyl chloroform measurements, Prinn et al. 2001 [JoC, SRC] ) inferred that global OH levels grew between 1979 and 1989, but then declined between 1989 and 2000, and also exhibited significant interannual variations. They concluded that these decadal global variations were driven principally by NH OH, with SH OH decreasing from 1979 to 1989 and staying essentially constant after that. Using the same AGAGE data and identical methyl chloroform emissions, a three-dimensional model analysis ( Krol and Lelieveld, 2003 [JoC] ) supported qualitatively (but not quantitatively) the earlier result ( Prinn et al., 2001 [JoC, SRC] ) that OH concentrations increased in the 1980 s and declined in the 1990 s. Prinn et al. 2001 [JoC, SRC] ) also estimated the emissions required to provide a zero trend in OH. These required methyl chloroform emissions differed substantially from industry estimates by ( McCulloch and Midgley 2001 ) ) particularly for 1996 to 2000 . However, Krol and Lelieveld 2003 [JoC] ) argued that the combination of possible underestimated recent emissions, especially the >20 Gg European emissions deduced by Krol et al. 2003 [JoC] ), and the recent decreasing effectiveness of the stratosphere as a sink for tropospheric methyl chloroform, may be sufficient to yield a zero deduced OH trend. As discussed in Section 2.3.4 ,estimates of European emissions by Reimann et al. 2005 [JoC] ) are an order of magnitude less than those of Krol et al. 2003 [JoC] ). In addition, Prinn et al. 2005a [JoC, SRC] ) extended the OH estimates through 2004 and showed that the Prinn et al. 2001 [JoC, SRC] ) decadal and interannual OH estimates remain valid even after accounting for the additional recent methyl chloroform emissions discussed in Section 2.3.4 .They also reconfirmed the OH maximum around 1989 and a larger OH minimum around 1998, with OH concentrations then recovering so that in 2003 they were comparable to those in 1979 . They noted that the 1997 to 1999 OH minimum coincides with, and is likely caused by, major global wildfires and an intense El Niño at that time. The 1997 Indonesian fires alone have been estimated to have lowered global late- 1997 OH levels by 6% due to carbon monoxide (CO) enhancements ( Duncan et al., 2003 [JoC] ).
Methyl chloroform is also destroyed in the stratosphere. Because its stratospheric loss frequency is less than that in the troposphere, the stratosphere becomes a less effective sink for tropospheric methyl chloroform over time ( Krol and Lelieveld, 2003 [JoC] ), and even becomes a small source to the troposphere beginning in 1999 in the reference case in the Prinn et al. 2001 [JoC, SRC] , 2005a ) model. Loss to the ocean has usually been considered irreversible, and its rates and uncertainties have been obtained from observations ( Yvon-Lewis and Butler, 2002 [JoC] ). However, Wennberg et al. 2004 [JoC] ) recently proposed that the polar oceans may have effectively stored methyl chloroform during the pre- 1992 years when its atmospheric levels were rising, but began re-emitting it in subsequent years, thus reducing the overall oceanic sink. Prinn et al. 2005a [JoC, SRC] ) tried both approaches and found that their inferred interannual and decadal OH variations were present using either formulation, but inferred OH was lower in the pre- 1992 years and higher after that using the Wennberg et al. 2004 [JoC] ) formulation.
More recently, Bousquet et al. 2005 [ARC] ) used an inverse method with a three-dimensional model and methyl chloroform measurements and concluded that substantial year-to-year variations occurred in global average OH concentrations between 1980 and 2000 . This conclusion was previously reached by Prinn et al. 2001 [JoC, SRC] ), but subsequently challenged by Krol and Lelieveld 2003 [JoC] ) who argued that these variations were caused by model shortcomings and that models need, in particular, to include observationally-based, interannually varying meteorology to provide accurate annual OH estimates. Neither the two-dimensional Prinn et al. 2001 [JoC, SRC] ) nor the three-dimensional Krol et al. 2003 [JoC] ) inversion models used interannually varying circulation. However, the Bousquet et al. 2005 [ARC] ) analysis, which uses observationally based meteorology and estimates OH on monthly time scales, yields interannual OH variations that agree very well with the Prinn et al. 2001 [JoC, SRC] ) and equivalent Krol and Lelieveld 2003 [JoC] ) estimates (see Figure 2.8 ). However, when Bousquet et al. 2005 [ARC] ) estimated both OH concentrations and methyl chloroform emissions (constrained by their uncertainties as reported by ( McCulloch and Midgley, 2001 ) ), the OH variations were reduced by 65% (dashed line in Figure 2.8 ). The error bars on the Prinn et al. 2001 [JoC, SRC] , 2005a ) OH estimates, which account for these emission uncertainties using Monte Carlo ensembles of inversions, also easily allow such a reduction in OH variability (thin vertical bars in Figure 2.8 ). This implies that these interannual OH variations are real, but only their phasing and not their amplitude, is well defined. Bousquet et al. 2005 [ARC] ) also deduced that OH in the SH shows a zero to small negative trend, in qualitative agreement with Prinn et al. 2001 [JoC, SRC] ). Short-term variations in OH were also recently deduced by Manning et al. 2005 [JoC, ARC] ) using 13 years of 14CO measurements in New Zealand and Antarctica. They found no significant long-term trend between 1989 and 2003 in SH OH but provided evidence for recurring multi-month OH variations of around 10%. They also deduced even larger (20%) OH decreases in 1991 and 1997, perhaps triggered by the 1991 Mt. Pinatubo eruption and the 1997 Indonesian fires. The similarity of many of these results to those from methyl chloroform discussed above is very important, given the independence of the two approaches.
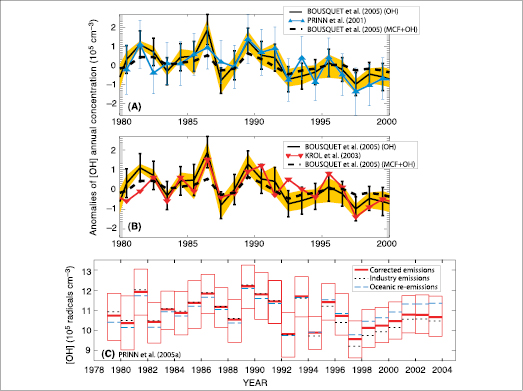
Figure 2.8. Estimates used to evaluate trends in weighted global average OH concentrations. (A) and (B): comparison of 1980 to 1999 OH anomalies (relative to their long-term means) inferred by Bousquet et al. 2005 [ARC] Prinn et al. 2001 [JoC, SRC] and Krol et al. 2003 [JoC] ) from AGAGE methyl chloroform observations, and by Bousquet et al. 2005 [ARC] ) when methyl chloroform emissions as well as OH are inferred; error bars for Bousquet et al. 2005 [ARC] ) refer to 1 standard deviation inversion errors while yellow areas refer to the envelope of their 18 OH inversions. (C) OH concentrations for 1979 to 2003 inferred by Prinn et al. 2005a [JoC, SRC] ) (utilising industry emissions corrected using recent methyl chloroform observations), showing the recovery of 2003 OH levels to 1979 levels; also shown are results assuming uncorrected emissions and estimates of recent oceanic re-emissions. Error bars in Prinn et al. 2001 [JoC, SRC] , 2005a ) are 1 standard deviation and include inversion, model, emission and calibration errors from large Monte Carlo ensembles (see Section 2.3.5 for details and references).
RF calculations of the LLGHGs are calculated from observed trends in the LLGHG concentrations and therefore OH concentrations do not directly affect them. Nevertheless OH trends are needed to quantify LLGHG budgets( Section 7.4 )and for understanding future trends in the LLGHGs and tropospheric ozone.
2.3.6 Ozone
In the TAR, separate estimates for RF due to changes in tropospheric and stratospheric ozone were given. Stratospheric ozone RF was derived from observations of ozone change from roughly 1979 to 1998 . Tropospheric ozone RF was based on chemical model results employing changes in precursor hydrocarbons, CO and nitrogen oxides (NOx). Over the satellite era (since approximately 1980 ), stratospheric ozone trends have been primarily caused by the Montreal Protocol gases, and in Ramaswamy et al. 2001 [NPR, MoS, SRC] ) the stratospheric ozone RF was implicitly attributed to these gases. Studies since then have investigated a number of possible causes of ozone change in the stratosphere and troposphere and the attribution of ozone trends to a given precursor is less clear. Nevertheless, stratospheric ozone and tropospheric ozone RFs are still treated separately in this report. However, the RFs are more associated with the vertical location of the ozone change than they are with the agent(s) responsible for the change.
2.3.6.1 Stratospheric Ozone
The TAR reported that ozone depletion in the stratosphere had caused a negative RF of –0.15 Wm–2 as a best estimate over the period since 1750 . A number of recent reports have assessed changes in stratospheric ozone and the research into its causes, including Chapters 3 and 4 of the 2002 Scientific Assessment of Ozone Depletion ( WMO, 2003 [NPR] ) and Chapter1 of IPCC/TEAP ( 2005 ). This section summarises the material from these reports and updates the key results using more recent research.
Global ozone amounts decreased between the late 1970 s and early 1990 s, with the lowest values occurring during 1992 to 1993 (roughly 6% below the 1964 to 1980 average), and slightly increasing values thereafter. Global ozone for the period 2000 to 2003 was approximately 4% below the 1964 to 1980 average values. Whether or not recently observed changes in ozone trends ( Newchurch et al., 2003 [JoC] ; Weatherhead and Andersen, 2006 [JoC] ) are already indicative of recovery of the global ozone layer is not yet clear and requires more detailed attribution of the drivers of the changes ( Steinbrecht et al., 2004a [JoC] (see also comment and reply: Cunnold et al., 2004 [JoC] and Steinbrecht et al., 2004b [JoC] Hadjinicolaou et al., 2005 [JoC, MoS] ; Krizan and Lastovicka, 2005; Weatherhead and Andersen, 2006 [JoC] ). The largest ozone changes since 1980 have occurred during the late winter and spring over Antarctica where average total column ozone in September and October is about 40 to 50% below pre- 1980 values ( WMO, 2003 [NPR] ). Ozone decreases over the Arctic have been less severe than have those over the Antarctic, due to higher temperature in the lower stratosphere and thus fewer polar stratospheric clouds to cause the chemical destruction. Arctic stratospheric ozone levels are more variable due to interannual variability in chemical loss and transport.
The temporally and seasonally non-uniform nature of stratospheric ozone trends has important implications for the resulting RF. Global ozone decreases result primarily from changes in the lower stratospheric extratropics. Total column ozone changes over the mid-latitudes of the SH are significantly larger than over the mid-latitudes of the NH. Averaged over the period 2000 to 2003, SH values are 6% below pre- 1980 values, while NH values are 3% lower. There is also significant seasonality in the NH ozone changes, with 4% decreases in winter to spring and 2% decreases in summer, while long-term SH changes are roughly 6% year round ( WMO, 2003 [NPR] ). Southern Hemisphere mid-latitude ozone shows significant decreases during the mid- 1980 s and essentially no response to the effects of the Mt. Pinatubo volcanic eruption in June 1991; both of these features remain unexplained. Pyle et al. 2005 [NPR] and Chipperfield et al. 2003 [NPR, MoS, SRC] ) assessed several studies that show that a substantial fraction (roughly 30%) of NH mid-latitude ozone trends are not directly attributable to anthropogenic chemistry, but are related to dynamical effects, such as tropopause height changes. These dynamical effects are likely to have contributed a larger fraction of the ozone RF in the NH mid-latitudes. The only study to assess this found that 50% of the RF related to stratospheric ozone changes between 20°N to 60°N over the period 1970 to 1997 is attributable to dynamics ( Forster and Tourpali, 2001 [JoC, MoS, SRC] ). These dynamical changes may well have an anthropogenic origin and could even be partly caused by stratospheric ozone changes themselves through lower stratospheric temperature changes ( Chipperfield et al., 2003 [NPR, MoS, SRC] ; Santer et al., 2004 [PoC, JoC, ARC] ), but are not directly related to chemical ozone loss.
At the time of writing, no study has utilised ozone trend observations after 1998 to update the RF values presented in Ramaswamy et al. 2001 [NPR, MoS, SRC] ). However, Hansen et al. 2005 [JoC, MoS, ARC] ) repeated the RF calculation based on the same trend data set employed by studies assessed in Ramaswamy et al. 2001 [NPR, MoS, SRC] ) and found an RF of roughly –0.06 Wm–2.A considerably stronger RF of –0.2 ± 0.1 Wm–2 previously estimated by the same group affected the Ramaswamy et al. 2001 [NPR, MoS, SRC] ) assessment. The two other studies assessed in Ramaswamy et al. 2001 [NPR, MoS, SRC] ), using similar trend data sets, found RFs of –0.01 Wm–2 and –0.10 Wm–2.Using the three estimates gives a revision of the observationally based RF for 1979 to 1998 to about –0.05 ± 0.05 Wm–2.
Gauss et al. 2006 [MoS] ) compared results from six chemical transport models that included changes in ozone precursors to simulate both the increase in the ozone in the troposphere and the ozone reduction in the stratosphere over the industrial era. The 1850 to 2000 annually averaged global mean stratospheric ozone column reduction for these models ranged between 14 and 29 Dobson units (DU). The overall pattern of the ozone changes from the models were similar but the magnitude of the ozone changes differed. The models showed a reduction in the ozone at high latitudes, ranging from around 20 to 40% in the SH and smaller changes in the NH. All models have a maximum ozone reduction around 15 km at high latitudes in the SH. Differences between the models were also found in the tropics, with some models simulating about a 10% increase in the lower stratosphere and other models simulating decreases. These differences were especially related to the altitude where the ozone trend switched from an increase in the troposphere to a decrease in the stratosphere, which ranged from close to the tropopause to around 27 km. Several studies have shown that ozone changes in the tropical lower stratosphere are very important for the magnitude and sign of the ozone RF ( Ramaswamy et al., 2001 [NPR, MoS, SRC] ). The resulting stratospheric ozone RF ranged between –0.12 and +0.07 Wm–2.Note that the models with either a small negative or a positive RF also had a small increase in tropical lower stratospheric ozone, resulting from increases in tropospheric ozone precursors; most of this increase would have occurred before the time of stratospheric ozone destruction by the Montreal Protocol gases. These RF calculations also did not include any negative RF that may have resulted from stratospheric water vapour increases. It has been suggested ( Shindell and Faluvegi, 2002 [MoS] ) that stratospheric ozone during 1957 to 1975 was lower by about 7 DU relative to the first half of the 20th century as a result of possible stratospheric water vapour increases; however, these long-term increases in stratospheric water vapour are uncertain (see Sections 2.3.7 and 3.4 ).
The stratospheric ozone RF is assessed to be –0.05 ± 0.10 Wm–2 between pre-industrial times and 2005 . The best estimate is from the observationally based 1979 to 1998 RF of –0.05 ± 0.05 Wm–2,with the uncertainty range increased to take into account ozone change prior to 1979, using the model results of Gauss et al. 2006 [MoS] ) as a guide. Note that this estimate takes into account causes of stratospheric ozone change in addition to those due to the Montreal Protocol gases. The level of scientific understanding is medium, unchanged from the TAR (see Section 2.9 , Table 2.11 ).
2.3.6.2 Tropospheric Ozone
The TAR identified large regional differences in observed trends in tropospheric ozone from ozonesondes and surface observations. The TAR estimate of RF from tropospheric ozone was +0.35 ± 0.15 Wm–2.Due to limited spatial and temporal coverage of observations of tropospheric ozone, the RF estimate is based on model simulations. In the TAR, the models considered only changes in the tropospheric photochemical system, driven by estimated emission changes (NOx,CO, non-methane volatile organic compounds (NMVOCs), and CH4)since pre-industrial times. Since the TAR, there have been major improvements in models. The new generation models include several Chemical Transport Models (CTMs) that couple stratospheric and tropospheric chemistry, as well as GCMs with on-line chemistry (both tropospheric and stratospheric). While the TAR simulations did not consider changes in ozone within the troposphere caused by reduced influx of ozone from the stratosphere (due to ozone depletion in the stratosphere), the new models include this process ( Gauss et al., 2006 [MoS] ). This advancement in modelling capabilities and the need to be consistent with how the RF due to changes in stratospheric ozone is derived (based on observed ozone changes) have led to a change in the definition of RF due to tropospheric ozone compared with that in the TAR. Changes in tropospheric ozone due to changes in transport of ozone across the tropopause, which are in turn caused by changes in stratospheric ozone, are now included.
Trends in anthropogenic emissions of ozone precursors for the period 1990 to 2000 have been compiled by the Emission Database for Global Atmospheric Research (EDGAR) consortium ( Olivier and Berdowski, 2001 [NPR] updated). For specific regions, there is significant variability over the period due to variations in the emissions from open biomass burning sources. For all components (NOx,CO and volatile organic compounds (VOCs)) industrialised regions like the USA and Organisation for Economic Co-operation and Development (OECD) Europe show reductions in emissions, while regions dominated by developing countries show significant growth in emissions. Recently, the tropospheric burdens of CO and NO2 were estimated from satellite observations ( Edwards et al., 2004 [JoC] ; Richter et al., 2005 [JoC] ), providing much needed data for model evaluation and very valuable constraints for emission estimates.
Assessment of long-term trends in tropospheric ozone is difficult due to the scarcity of representative observing sites with long records. The long-term tropospheric ozone trends vary both in terms of sign and magnitude and in the possible causes for the change ( (Oltmans et al., 2006 ) ). Trends in tropospheric ozone at northern middle and high latitudes have been estimated based on ozonesonde data by WMO 2003 [NPR] Naja et al. 2003 [JoC] Naja and Akimoto 2004 [JoC] Tarasick et al. 2005 [JoC] ( and Oltmans et al. 2006 ) ). Over Europe, ozone in the free troposphere increased from the early 20th century until the late 1980 s; since then the trend has levelled off or been slightly negative. Naja and Akimoto 2004 [JoC] ) analysed 33 years of ozonesonde data from Japanese stations, and showed an increase in ozone in the lower troposphere (750–550 hPa) between the periods 1970 to 1985 and 1986 to 2002 of 12 to 15% at Sapporo and Tsukuba (43°N and 36°N) and 35% at Kagoshima (32°N). Trajectory analysis indicates that the more southerly station, Kagoshima, is significantly more influenced by air originating over China, while Sapporo and Tsukuba are more influenced by air from Eurasia. At Naha (26°N) a positive trend (5% per decade) is found between 700 and 300 hPa ( 1990 – 2004 ), while between the surface and 700 hPa a slightly negative trend is observed ( (Oltmans et al., 2006 ) ). Ozonesondes from Canadian stations show negative trends in tropospheric ozone between 1980 and 1990, and a rebound with positive trends during 1991 to 2001 ( Tarasick et al., 2005 [JoC] ). Analysis of stratosphere-troposphere exchange processes indicates that the rebound during the 1990 s may be partly a result of small changes in atmospheric circulation.
Trends are also derived from surface observations. Jaffe et al. 2003 [JoC] ) derived a positive trend of 1.4% yr–1 between 1988 and 2003 using measurements from Lassen Volcanic Park in California (1,750 m above sea level), consistent with the trend derived by comparing two aircraft campaigns ( Parrish et al., 2004 [JoC] ). However, a number of other sites show insignificant changes over the USA over the last 15 years ( (Oltmans et al., 2006 ) ). Over Europe and North America, observations from Whiteface Mountain, Wallops Island, Hohenpeisenberg, Zugspitze and Mace Head (flow from the European sector) show small trends or reductions during summer, while there is an increase during winter ( (Oltmans et al., 2006 ) ). These observations are consistent with reduced NOx emissions ( (Jonson et al., 2005 ) ). North Atlantic stations (Mace Head, Izana and Bermuda) indicate increased ozone ( (Oltmans et al., 2006 ) ). Over the North Atlantic (40°N–60°N) measurements from ships ( Lelieveld et al., 2004 [JoC] ) show insignificant trends in ozone, however, at Mace Head a positive trend of 0.49 ± 0.19 ppb yr–1 for the period 1987 to 2003 is found, with the largest contribution from air coming from the Atlantic sector ( Simmonds et al., 2004 [SRC] ).
In the tropics, very few long-term ozonesonde measurements are available. At Irene in South Africa (26°S), Diab et al. 2004 [JoC] ) found an increase between the 1990 to 1994 and 1998 to 2002 periods of about 10 ppb close to the surface (except in summer) and in the upper troposphere during winter. Thompson et al. 2001 [JoC] ) found no significant trend during 1979 to 1992, based on Total Ozone Mapping Spectrometer (TOMS) satellite data. More recent observations ( 1994 to 2003, in situ data from the Measurement of Ozone by Airbus In-service Aircraft (MOZAIC) program) show significant trends in free-tropospheric ozone (7.7 to 11.3 km altitude) in the tropics: 1.12 ± 0.05 ppb yr–1 and 1.03 ± 0.08 ppb yr–1 in the NH tropics and SH tropics, respectively ( Bortz and Prather, 2006 [JoC, ARC] ). Ozonesonde measurements over the southwest Pacific indicate an increased frequency of near-zero ozone in the upper troposphere, suggesting a link to an increased frequency of deep convection there since the 1980 s ( Solomon et al., 2005 [PoC, JoC, ARC] ).
At southern mid-latitudes, surface observations from Cape Point, Cape Grim, the Atlantic Ocean (from ship) and from sondes at Lauder (850–700 hPa) show positive trends in ozone concentrations, in particular during the biomass burning season in the SH ( (Oltmans et al., 2006 ) ). However, the trend is not accompanied by a similar trend in CO, as expected if biomass burning had increased. The increase is largest at Cape Point, reaching 20% per decade (in September). At Lauder, the increase is confined to the lower troposphere.
Changes in tropospheric ozone and the corresponding RF have been estimated in a number of recent model studies ( Hauglustaine and Brasseur, 2001 [JoC, MoS, ARC] ; Mickley et al., 2001 [JoC, MoS, SRC] ; Shindell et al., 2003a [MoS] ; Mickley et al., 2004 [JoC, MoS, SRC] ; Wong et al., 2004 [JoC, MoS] ; Liao and Seinfeld, 2005 [JoC, MoS] ; Shindell et al., 2005 [PoC, JoC, MoS, ARC] ). In addition, a multi-model experiment including 10 global models was organised through the Atmospheric Composition Change: an European Network (ACCENT; Gauss et al., 2006 [MoS] ). Four of the ten ACCENT models have detailed stratospheric chemistry. The adjusted RF for all models was calculated by the same radiative transfer model. The normalised adjusted RF for the ACCENT models was +0.032 ± 0.006 Wm–2 DU–1,which is significantly lower than the TAR estimate of +0.042 Wm–2 DU–1.
The simulated RFs for tropospheric ozone increases since 1750 are shown in Figure 2.9 .Most of the calculations used the same set of assumptions about pre-industrial emissions (zero anthropogenic emissions and biomass burning sources reduced by 90%). Emissions of NOx from soils and biogenic hydrocarbons were generally assumed to be natural and were thus not changed (see, e.g., Section 7.4 ). In one study ( Hauglustaine and Brasseur, 2001 [JoC, MoS, ARC] ), pre-industrial NOx emissions from soils were reduced based on changes in the use of fertilizers. Six of the ACCENT models also made coupled climate-chemistry simulations including climate change since pre-industrial times. The difference between the RFs in the coupled climate-chemistry and the chemistry-only simulations, which indicate the possible climate feedback to tropospheric ozone, was positive in all models but generally small( Figure 2.9 ).
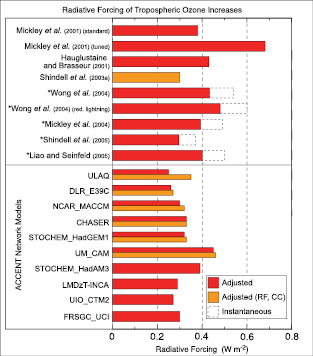
Figure 2.9. Calculated RF due to tropospheric ozone change since pre-industrial time based on CTM and GCM model simulations published since the TAR. Estimates with GCMs including the effect of climate change since 1750 are given by orange bars (Adjusted RF, CC). Studies denoted with an (*) give only the instantaneous RF in the original publications. Stratospheric-adjusted RFs for these are estimated by reducing the instantaneous RF (indicated by the dashed bars) by 20%. The instantaneous RF from Mickley et al. 2001 [JoC, MoS, SRC] ) is reported as an adjusted RF in Gauss et al. 2006 [MoS] ). ACCENT models include ULAQ: University of L’Aquila; DLR_E39C: Deutsches Zentrum für Luft- und Raumfahrt European Centre Hamburg Model; NCAR_MACCM: National Center for Atmospheric Research Middle Atmosphere Community Climate Model; CHASER: Chemical Atmospheric GCM for Study of Atmospheric Environment and Radiative Forcing; STOCHEM_HadGEM1: United Kingdom Meteorological Office global atmospheric chemistry model /Hadley Centre Global Environmental Model 1; UM_CAM: United Kingdom Meteorological Office Unified Model GCM with Cambridge University chemistry; STOCHEM_HadAM3: United Kingdom Meteorological Office global atmospheric chemistry model/Hadley Centre Atmospheric Model; LMDzT-INCA: Laboratoire de Météorologie Dynamique GCM-INteraction with Chemistry and Aerosols; UIO_CTM2: University of Oslo CTM; FRSGC_UCI: Frontier Research System for Global Change/University of California at Irvine CTM.
Ageneral feature of the models is their inability to reproduce the low ozone concentrations indicated by the very uncertain semi-quantitative observations (e.g., ( Pavelin et al., 1999 ) ) during the late 19th century. Mickley et al. 2001 [JoC, MoS, SRC] ) tuned their model by reducing pre-industrial lightning and soil sources of NOx and increasing natural NMVOC emissions to obtain close agreement with the observations. The ozone RF then increased by 50 to 80% compared to their standard calculations. However, there are still several aspects of the early observations that the tuned model did not capture.
The best estimate for the RF of tropospheric ozone increases is +0.35 Wm–2,taken as the median of the RF values in Figure 2.9 (adjusted and non-climate change values only, i.e., the red bars). The best estimate is unchanged from the TAR. The uncertainties in the estimated RF by tropospheric ozone originate from two factors: the models used (CTM/GCM model formulation, radiative transfer models), and the potential overestimation of pre-industrial ozone levels in the models. The 5 to 95% confidence interval, assumed to be represented by the range of the results in Figure 2.9 ,is +0.25 to +0.65 Wm–2.A medium level of scientific understanding is adopted, also unchanged from the TAR (see Section 2.9 , Table 2.11 ).
2.3.7 Stratospheric Water Vapour
The TAR noted that several studies had indicated long-term increases in stratospheric water vapour and acknowledged that these trends would contribute a significant radiative impact. However, it only considered the stratospheric water vapour increase expected from CH4 increases as an RF, and this was estimated to contribute 2 to 5% of the total CH4 RF (about +0.02 Wm–2).
Section 3.4 discusses the evidence for stratospheric water vapour trends and presents the current understanding of their possible causes. There are now 14 years of global stratospheric water vapour measurements from Halogen Occultation Experiment (HALOE) and continued balloon-based measurements (since 1980 ) at Boulder, Colorado. There is some evidence of a sustained long-term increase in stratospheric water vapour of around 0.05 ppm yr–1 from 1980 until roughly 2000, since then water vapour concentrations in the lower stratosphere have been decreasing (see Section 3.4 for details and references). As well as CH4 increases, several other indirect forcing mechanisms have been proposed, including: a) volcanic eruptions (Considi ne et al., 2001; Joshi and Shine, 2003 [JoC] ); b) biomass burning aerosol ( Sherwood, 2002 [JoC, ARC] ); c) tropospheric (SO2 ;Notholt et al., 2005 [JoC] ) and d) changes in CH4 oxidation rates from changes in stratospheric chlorine, ozone and OH ( (Rockmann et al., 2004 ) ). These are mechanisms that can be linked to an external forcing agent. Other proposed mechanisms are more associated with climate feedbacks and are related to changes in tropopause temperatures or circulation ( Stuber et al., 2001a [JoC, SRC] ; Fueglistaler et al., 2004 [JoC] ). From these studies, there is little quantification of the stratospheric water vapour change attributable to different causes. It is also likely that different mechanisms are affecting water vapour trends at different altitudes.
Since the TAR, several further calculations of the radiative balance change due to changes in stratospheric water vapour have been performed ( Forster and Shine, 1999 [JoC, SRC] ; Oinas et al., 2001 [JoC] ; Shindell, 2001 [JoC] ; Smith et al., 2001 [JoC, MoS, ARC] ; Forster and Shine, 2002 [JoC, SRC] Smith et al. 2001 [JoC, MoS, ARC] ) estimated a +0.12 to +0.2 Wm–2 per decade range for the RF from the change in stratospheric water vapour, using HALOE satellite data. Shindell 2001 [JoC] ) estimated an RF of about +0.2 Wm–2 in a period of two decades, using a GCM to estimate the increase in water vapour in the stratosphere from oxidation of CH4 and including climate feedback changes associated with an increase in greenhouse gases. Forster and Shine 2002 [JoC, SRC] ) used a constant 0.05 ppm yr–1 trend in water vapour at pressures of 100 to 10 hPa and estimated the RF to be +0.29 Wm–2 for 1980 to 2000 . GCM radiation codes can have a factor of two uncertainty in their modelling of this RF ( Oinas et al., 2001 [JoC] ). For the purposes of this chapter, the above RF estimates are not readily attributable to forcing agent(s) and uncertainty as to the causes of the observed change precludes all but the component due to CH4 increases being considered a forcing. Two related CTM studies have calculated the RF associated with increases in CH4 since pre-industrial times ( Hansen and Sato, 2001 [JoC, MoS, ARC] ; Hansen et al., 2005 [JoC, MoS, ARC] ), but no dynamical feedbacks were included in those estimates. Hansen et al. 2005 [JoC, MoS, ARC] ) estimated an RF of +0.07 ± 0.01 Wm–2 for the stratospheric water vapour changes over 1750 to 2000, which is at least a factor of three larger than the TAR value. The RF from direct injection of water vapour by aircraft is believed to be an order of magnitude smaller than this, at about +0.002 Wm–2 ( IPCC, 1999 [NPR] ). There has been little trend in CH4 concentration since 2000 (see Section 2.3.2 ); therefore the best estimate of the stratospheric water vapour RF from CH4 oxidation (+0.07 Wm–2)is based on the Hansen et al. 2005 [JoC, MoS, ARC] ) calculation. The 90% confidence range is estimated as ±0.05 Wm–2,from the range of the RF studies that included other effects. There is a low level of scientific understanding in this estimate, as there is only a partial understanding of the vertical profile of CH4-induced stratospheric water vapour change( Section 2.9 , Table 2.11 ). Other human causes of stratospheric water vapour change are unquantified and have a very low level of scientific understanding.
2.3.8 Observations of Long-Lived Greenhouse Gas Radiative Effects
Observations of the clear-sky radiation emerging at the top of the atmosphere and at the surface have been conducted. Such observations, by their nature, do not measure RF as defined here. Instead, they yield a perspective on the influence of various species on the transfer of radiation in the atmosphere. Most importantly, the conditions involved with these observations involve varying thermal and moisture profiles in the atmosphere such that they do not conform to the conditions underlying the RF definition (see Section 2.2 ). There is a more comprehensive discussion of observations of the Earth’s radiative balance in Section 3.4 .
Harries et al. 2001 [JoC, MoS] ) analysed spectra of the outgoing longwave radiation as measured by two satellites in 1970 and 1997 over the tropical Pacific Ocean. The reduced brightness temperature observed in the spectral regions of many of the greenhouse gases is experimental evidence for an increase in the Earth’s greenhouse effect. In particular, the spectral signatures were large for CO2 and CH4.The halocarbons, with their large change between 1970 and 1997, also had an impact on the brightness temperature. Philipona et al. 2004 [JoC, MoS] ) found an increase in the measured longwave downward radiation at the surface over the period from 1995 to 2002 at eight stations over the central Alps. A significant increase in the clear-sky longwave downward flux was found to be due to an enhanced greenhouse effect after combining the measurements with model calculations to estimate the contribution from increases in temperature and humidity. While both types of observations attest to the radiative influences of the gases, they should not be interpreted as having a direct linkage to the value of RFs in Section 2.3 .
2.4 Aerosols
2.4.1 Introduction and Summary of the Third Assessment Report
The TAR categorised aerosol RFs into direct and indirect effects. The direct effect is the mechanism by which aerosols scatter and absorb shortwave and longwave radiation, thereby altering the radiative balance of the Earth-atmosphere system. Sulphate, fossil fuel organic carbon, fossil fuel black carbon, biomass burning and mineral dust aerosols were all identified as having a significant anthropogenic component and exerting a significant direct RF. Key parameters for determining the direct RF are the aerosol optical properties (the single scattering albedo, ωo,specific extinction coefficient,ke and the scattering phase function), which vary as a function of wavelength and relative humidity, and the atmospheric loading and geographic distribution of the aerosols in the horizontal and vertical, which vary as a function of time (e.g., Haywood and Boucher, 2000 [JoC, MoS, SRC] ; Penner et al., 2001 [NPR, ARC] ; Ramaswamy et al., 2001 [NPR, MoS, SRC] ). Scattering aerosols exert a net negative direct RF, while partially absorbing aerosols may exert a negative top-of-the-atmosphere (TOA) direct RF over dark surfaces such as oceans or dark forest surfaces, and a positive TOA RF over bright surfaces such as desert, snow and ice, or if the aerosol is above cloud (e.g., Chylek and Wong, 1995 [JoC] ; Haywood and Shine, 1995 [JoC, SRC] ). Both positive and negative TOA direct RF mechanisms reduce the shortwave irradiance at the surface. The longwave direct RF is only substantial if the aerosol particles are large and occur in considerable concentrations at higher altitudes (e.g., Tegen et al., 1996 [JoC, MoS, ARC] ). The direct RF due to tropospheric aerosols is most frequently derived at TOA rather than at the tropopause because shortwave radiative transfer calculations have shown a negligible difference between the two (e.g., Haywood and Shine, 1997 [MoS, SRC] ; Section 2.2 ). The surface forcing will be approximately the same as the direct RF at the TOA for scattering aerosols, but for partially absorbing aerosols the surface forcing may be many times stronger than the TOA direct RF (e.g., Ramanathan et al., 2001b [JoC, MoS, SRC] and references therein).
The indirect effect is the mechanism by which aerosols modify the microphysical and hence the radiative properties, amount and lifetime of clouds( Figure 2.10 ). Key parameters for determining the indirect effect are the effectiveness of an aerosol particle to act as a cloud condensation nucleus, which is a function of the size, chemical composition, mixing state and ambient environment (e.g., Penner et al., 2001 [NPR, ARC] ). The microphysically induced effect on the cloud droplet number concentration and hence the cloud droplet size, with the liquid water content held fixed has been called the ‘first indirect effect’ (e.g., Ramaswamy et al., 2001 [NPR, MoS, SRC] ), the ‘cloud albedo effect’ (e.g., Lohmann and Feichter, 2005 [SRC] ), or the ‘Twomey effect’ (e.g., Twomey, 1977 [JoC] ). The microphysically induced effect on the liquid water content, cloud height, and lifetime of clouds has been called the ‘second indirect effect’ (e.g., Ramaswamy et al., 2001 [NPR, MoS, SRC] ), the ‘cloud lifetime effect’ (e.g., Lohmann and Feichter, 2005 [SRC] ) or the ‘Albrecht effect’ (e.g., Albrecht, 1989 [JoC] ). The TAR split the indirect effect into the first indirect effect, and the second indirect effect. Throughout this report, these effects are denoted as ‘cloud albedo effect’ and ‘cloud lifetime effect’, respectively, as these terms are more descriptive of the microphysical processes that occur. The cloud albedo effect was considered in the TAR to be an RF because global model calculations could be performed to describe the influence of increased aerosol concentration on the cloud optical properties while holding the liquid water content of the cloud fixed (i.e., in an entirely diagnostic manner where feedback mechanisms do not occur). The TAR considered the cloud albedo effect to be a key uncertainty in the RF of climate but did not assign a best estimate of the RF, and showed a range of RF between 0 and –2 Wm–2 in the context of liquid water clouds. The other indirect effects were not considered to be RFs because, in suppressing drizzle, increasing the cloud height or the cloud lifetime in atmospheric models( Figure 2.10 ), the hydrological cycle is invariably altered (i.e., feedbacks occur; see Section 7.5 ). The TAR also discussed the impact of anthropogenic aerosols on the formation and modification of the physical and radiative properties of ice clouds ( Penner et al., 2001 [NPR, ARC] ), although quantification of an RF from this mechanism was not considered appropriate given the host of uncertainties and unknowns surrounding ice cloud nucleation and physics.
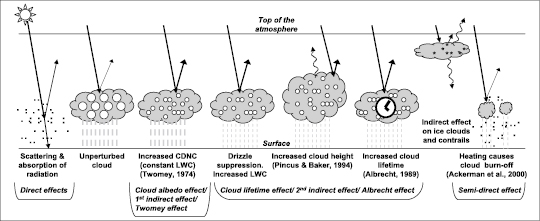
Figure 2.10. Schematic diagram showing the various radiative mechanisms associated with cloud effects that have been identified as significant in relation to aerosols (modified from Haywood and Boucher, 2000 [JoC, MoS, SRC] ). The small black dots represent aerosol particles; the larger open circles cloud droplets. Straight lines represent the incident and reflected solar radiation, and wavy lines represent terrestrial radiation. The filled white circles indicate cloud droplet number concentration (CDNC). The unperturbed cloud contains larger cloud drops as only natural aerosols are available as cloud condensation nuclei, while the perturbed cloud contains a greater number of smaller cloud drops as both natural and anthropogenic aerosols are available as cloud condensation nuclei (CCN). The vertical grey dashes represent rainfall, and LWC refers to the liquid water content.
The TAR did not include any assessment of the semi-direct effect (e.g., Hansen et al., 1997 [JoC, MoS, ARC] ; Ackerman et al., 2000a [JoC] ; Jacobson, 2002 [JoC] ; Menon et al., 2003 [JoC, MoS, ARC] ; Cook and Highwood, 2004 [JoC, MoS] ; Johnson et al., 2004 [JoC] ), which is the mechanism by which absorption of shortwave radiation by tropospheric aerosols leads to heating of the troposphere that in turn changes the relative humidity and the stability of the troposphere and thereby influences cloud formation and lifetime. In this report, the semi-direct effect is not strictly considered an RF because of modifications to the hydrological cycle, as discussed in Section 7.5 (see also Sections 2.2 , 2.8 and 2.4.5 ).
Since the TAR, there have been substantial developments in observations and modelling of tropospheric aerosols; these are discussed in turn in the following sections.
2.4.2 Developments Related to Aerosol Observations
Surface-based measurements of aerosol properties such as size distribution, chemical composition, scattering and absorption continue to be performed at a number of sites, either at long-term monitoring sites, or specifically as part of intensive field campaigns. These in situ measurements provide essential validation for global models, for example, by constraining aerosol concentrations at the surface and by providing high-quality information about chemical composition and local trends. In addition, they provide key information about variability on various time scales. Comparisons of in situ measurements against those from global atmospheric models are complicated by differences in meteorological conditions and because in situ measurements are representative of conditions mostly at or near the surface while the direct and indirect RFs depend on the aerosol vertical profile. For example, the spatial resolution of global model grid boxes is typically a few degrees of latitude and longitude and the time steps for the atmospheric dynamics and radiation calculations may be minutes to hours depending on the process to be studied; this poses limitations when comparing with observations conducted over smaller spatial extent and shorter time duration.
Combinations of satellite and surface-based observations provide near-global retrievals of aerosol properties. These are discussed in this subsection; the emissions estimates, trends and in situ measurements of the physical and optical properties are discussed with respect to their influence on RF in Section 2.4.4 .Further detailed discussions of the recent satellite observations of aerosol properties and a satellite-measurement based assessment of the aerosol direct RF are given by Yu et al. 2006 [MoS] ).
2.4.2.1 Satellite Retrievals
Satellite retrievals of aerosol optical depth in cloud-free regions have improved via new generation sensors ( Kaufman et al., 2002 [JoC, SRC] ) and an expanded global validation program ( Holben et al., 2001 [JoC] ). Advanced aerosol retrieval products such as aerosol fine-mode fraction and effective particle radius have been developed and offer potential for improving estimates of the aerosol direct radiative effect. Additionally, efforts have been made to determine the anthropogenic component of aerosol and associated direct RF, as discussed by Kaufman et al. 2002 [JoC, SRC] ) and implemented by Bellouin et al. 2005 [JoC, MoS, SRC] and Chung et al. 2005 [JoC, MoS, SRC] ). However, validation programs for these advanced products have yet to be developed and initial assessments indicate some systematic errors ( Levy et al., 2003 [JoC] ; Anderson et al., 2005a [JoC] ; Chu et al., 2005 [JoC] ), suggesting that the routine differentiation between natural and anthropogenic aerosols from satellite retrievals remains very challenging.
2.4.2.1.1 Satellite retrievals of aerosol optical depth
Figure 2.11 shows an example of aerosol optical depth τaer (mid-visible wavelength) retrieved over both land and ocean, together with geographical positions of aerosol instrumentation. Table 2.2 provides a summary of aerosol data currently available from satellite instrumentation, together with acronyms for the instruments. τaer from the Moderate Resolution Imaging Spectrometer (MODIS) instrument for the January to March 2001 average( Figure 2.11 ,top panel) clearly differs from that for the August to October 2001 average( Figure 2.11 ,bottom panel) ( Kaufman et al., 1997 [JoC] ; Tanré et al., 1997 [JoC] ). Seasonal variability in τaer can be seen; biomass burning aerosol is most strongly evident over the Gulf of Guinea in Figure 2.11 (top panel) but shifts to southern Africa in Figure 2.11 (bottom panel). Likewise, the biomass burning in South America is most evident in Figure 2.11 (bottom panel). In Figure 2.11 (top panel), transport of mineral dust from Africa to South America is discernible while in Figure 2.11 (bottom panel) mineral dust is transported over the West Indies and Central America. Industrial aerosol, which consists of a mixture of sulphates, organic and black carbon, nitrates and industrial dust, is evident over many continental regions of the NH. Sea salt aerosol is visible in oceanic regions where the wind speed is high (e.g., south of 45°S). The MODIS aerosol algorithm is currently unable to make routine retrievals over highly reflective surfaces such as deserts, snow cover, ice and areas affected by ocean glint, or over high-latitude regions when the solar insolation is insufficient.
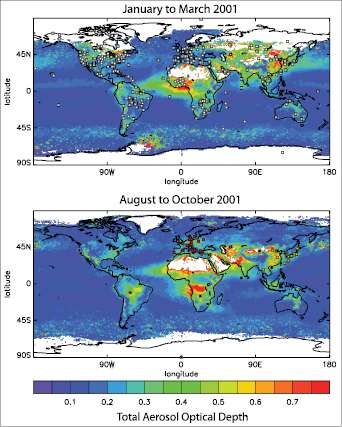
Figure 2.11. Aerosol optical depth, τaer,at 0.55 μm (colour bar) as determined by the MODIS instrument for the January to March 2001 mean (top panel) and for the August to October 2001 mean (bottom panel). The top panel also shows the location of AERONET sites (white squares) that have been operated (not necessary continuously) since 1996 . The bottom panel also shows the location of different aerosol lidar networks (red: EARLINET, orange: ADNET, black: MPLNET).
Table 2.2. Periods of operation, spectral bands and products available from various different satellite sensors that have been used to retrieve aerosol properties. Notes: a DRE is the direct radiative effect and includes both natural and anthropogenic sources (see Table 2.3 ). The Angstrom exponent, a, is the wavelength dependence of τaer and is defined by a = –ln(τaerλ1/τaerλ2)/ ln(λ1 / λ2)where λ1 = wavelength 1 and λ2 = wavelength 2. b TOMS followed up by the Ozone Monitoring Instrument (OMI) on the Earth Observing System (EOS) Aura satellite, launched July 2004 .
| Satellite Instrument | Period of Operation | Spectral Bands | Productsa | Comment and Reference |
|---|---|---|---|---|
| AVHRR (Advanced Very High Resolution Radiometer) a | 1979 to present | 5bands (0.63, 0.87, 3.7, 10.5 and 11.5 µm) | τaer,α | 1-channel retrieval gives τλ=0.63 over ocean (Husar et al., 1997; Ignatov andStowe, 2002)2-channel using 0.63 µm and 0.86 µm gives τλ=0.55 and α over ocean assuming mono-modal aerosol size distribution (Mishchenko et al., 1999)2-channel using 0.63 µm and 0.86 µm gives τλ=0.55 and α over dark forests and lake surfaces (Soufflet et al., 1997)2-channel using 0.64 µm and 0.83 µm gives τλ=0.55 and α over ocean assuming a bimodal aerosol size distribution (Higurashi and Nakajima, 1999; Higurashi et al., 2000) |
| TOMSb (Total Ozone Mapping Spectrometer) | 1979 to present | 0. 33 µm, 0.36 µm | Aerosol Index, τaer | Aerosol index to τaer conversion sensitive to the altitude of the 8 mono-modalaerosol models used in the retrieval (Torres et al., 2002). |
| POLDER (Polarization and Directionality of the Earth’s Reflectances) | Nov 1996 to June 1997; Apr 2003 to Oct 2003; Jan 2005 to present | 8bands (0.44 to 0.91 µm) | τaer,α, DRE | Multiple view angles and polarization capabilities.0.67 µm and 0.86 µm radiances used with 12 mono-modal aerosol models overocean (Goloub et al., 1999; Deuzé et al., 2000).Polarization allows fine particle retrieval over land (Herman et al., 1997;Goloub and Arino, 2000).DRE determined over ocean (Boucher and Tanré, 2000; Bellouin et al., 2003). |
| OCTS (Ocean Colour and Temperature Scanner) | Nov 1996 to Jun 1997; Apr 2003 to Oct 2003 | 9bands (0.41 to 0.86 µm) and 3.9 µm | τaer,α | 0.67 µm and 0.86 µm retrieval gives τλ=0.50 and α over ocean. Bi-modal aerosolsize distribution assumed (Nakajima and Higurashi, 1998; Higurashi et al., 2000). |
| MODIS (Moderate Resolution Imaging Spectrometer) | 2000 to present | 12 bands (0.41 to 2.1 µm) | τaer,α, DRE | Retrievals developed over ocean surfaces using bi-modal size distributions(Tanré et al., 1997; Remer et al., 2002).Retrievals developed over land except bright surfaces (Kaufman et al., 1997; Chu et al., 2002).Optical depth speciation and DRE determined over ocean and land (e.g., Bellouin et al., 2005; Kaufman et al., 2005a). |
| MISR (Multi-angle Imaging Spectro-Radiometer) | 2000 to present | 4bands (0.47 to 0.86 µm) | τaer,α | 9different viewing angles. Five climatological mixing groups composed of fourcomponent particles are used in the retrieval algorithm (Kahn et al., 2001; Kahnet al., 2005). Retrievals over bright surfaces are possible (Martonchik et al., 2004). |
| CERES (Clouds and the Earth’s Radiant Energy System) | 1998 to present | DRE | DRE determined by a regression of, for example, Visible Infrared Scanner (VIRS;AVHRR-like) τaer against upwelling irradiance (Loeb and Kato; 2002). | |
| GLAS (Geoscience Laser Altimeter System) | 2003 to present | Active lidar (0.53, 1.06 µm) | Aerosol vertical profile | Lidar footprint roughly 70 m at 170 m intervals. 8-day repeat orbiting cycle (Spinhirneet al., 2005). |
| ATSR-2/AATSR (Along Track Scanning Radiometer/Advanced ATSR) | 1996 to present | 4bands (0.56 to 1.65 µm) | τaer,α | Nadir and 52° forward viewing geometry. 40 aerosol climatological mixturescontaining up to six aerosol species are used in the retrievals (Veefkind et al., 1998; Holzer-Popp et al., 2002). |
| SeaWiFS (Sea-Viewing Wide Field-of-View Sensor) | 1997 to present | 0.765 and 0.865 µm (ocean) 0.41 to 0.67 µm (land) | τaer,α | 2-channel using 0.765 µm and 0.856 µm gives τλ=0.856 and α over ocean. Bi-modalaerosol size distribution assumed (M. Wang et al., 2005). Retrievals over land andocean using six visible channels from 0.41 to 0.67μm (von Hoyningen-Huene, 2003;Lee et al., 2004) also developed. |
Early retrievals for estimating τaer include the Advanced Very High Resolution Radiometer (AVHRR) single channel retrieval (e.g., Husar et al., 1997 [JoC] ; Ignatov and Stowe, 2002 [JoC, MoS] ), and the ultraviolet-based retrieval from the TOMS (e.g., Torres et al., 2002 [JoC] ). A dual channel AVHRR retrieval has also been developed (e.g., ( Mishchenko et al., 1999; ) Geogdzhayev et al., 2002 [JoC] ). Retrievals by the AVHRR are generally only performed over ocean surfaces where the surface reflectance characteristics are relatively well known, although retrievals are also possible over dark land surfaces such as boreal forests and lakes ( (Soufflet et al., 1997 ) ). The TOMS retrieval is essentially independent of surface reflectance thereby allowing retrievals over both land and ocean ( Torres et al., 2002 [JoC] ), but is sensitive to the altitude of the aerosol, and has a relatively low spatial resolution. While these retrievals only use a limited number of spectral bands and lack sophistication compared to those from dedicated satellite instruments, they have the advantage of offering continuous long-term data sets (e.g., Geogdzhayev et al., 2002 [JoC] ).
Early retrievals have been superseded by those from dedicated aerosol instruments (e.g., Kaufman et al., 2002 [JoC, SRC] ). Polarization and Directionality of the Earth’s Reflectance (POLDER) uses a combination of spectral channels (0.44–0.91 µm) with several viewing angles, and measures polarization of radiation. Aerosol optical depth and Ångstrom exponent (α) over ocean ( Deuzé et al., 2000 [JoC, MoS] ), τaer over land ( Deuzé et al., 2001 [JoC] ) and the direct radiative effect of aerosols ( Boucher and Tanré, 2000 [JoC, MoS, SRC] ; Bellouin et al., 2003 [JoC, SRC] ) have all been developed. Algorithms for aerosol retrievals using MODIS have been developed and validated over both ocean ( Tanré et al., 1997 [JoC] ) and land surfaces ( Kaufman et al., 1997 [JoC] ). The uncertainty in these retrievals of τaer is necessarily higher over land ( Chu et al., 2002 [JoC] ) than over oceans ( Remer et al., 2002 [JoC] ) owing to uncertainties in land surface reflectance characteristics, but can be minimised by careful selection of the viewing geometry ( Chylek et al., 2003 [JoC, MoS] ). In addition, new algorithms have been developed for discriminating between sea salt, dust or biomass burning and industrial pollution over oceans ( Bellouin et al., 2003 [JoC, SRC] , 2005; Kaufman et al., 2005a [JoC, MoS] ) that allow for a more comprehensive comparison against aerosol models. Multi-angle Imaging Spectro-Radiometer (MISR) retrievals have been developed using multiple viewing capability to determine aerosol parameters over ocean ( Kahn et al., 2001 [JoC] ) and land surfaces, including highly reflective surfaces such as deserts ( Martonchik et al., 2004 [JoC] ). Five typical aerosol climatologies, each containing four aerosol components, are used in the retrievals, and the optimum radiance signature is determined for nine viewing geometries and two different radiances. The results have been validated against those from the Aerosol RObotic NETwork (AERONET; see Section 2.4.3 ). Along Track Scanning Radiometer (ATSR) and ATSR-2 retrievals ( Veefkind et al., 1998 [JoC] ; Holzer-Popp et al., 2002 [JoC, MoS] ) use a relatively wide spectral range (0.56–1.65 µm), and two viewing directions and aerosol climatologies from the Optical Parameters of Aerosols and Clouds (OPAC) database ( Hess et al., 1998 [JoC] ) to make τaer retrievals over both ocean and land ( Robles-Gonzalez et al., 2000 [JoC] ). The Ocean Colour and Temperature Scanner (OCTS) retrieval has a basis similar to the dual wavelength retrieval from AVHRR and uses wavelengths over the range 0.41 to 0.86 µm to derive τaer and α over oceans (e.g., Higurashi et al., 2000 [JoC] ) using a bi-modal aerosol size distribution. The Sea-Viewing Wide Field-of-View Sensor (SeaWiFs) uses 0.765 µm and 0.856 µm radiances to provide τλ=0.856 and α over ocean using a bi-modal aerosol size distribution (M. Wang et al., 2005 [Ambiguous] ). Further SeaWiFs aerosol products have been developed over both land and ocean using six and eight visible channels, respectively (e.g., von Hoyningen-Heune et al., 2003; Lee et al., 2004 [JoC, MoS] ).
Despite the increased sophistication and realism of the aerosol retrieval algorithms, discrepancies exist between retrievals of τaer even over ocean regions (e.g., Penner et al., 2002 [JoC, MoS, ARC] ; Myhre et al., 2004a [JoC, SRC] , 2005b; Jeong et al., 2005 [JoC] ; Kinne et al., 2006 [MoS, SRC] ). These discrepancies are due to different assumptions in the cloud clearing algorithms, aerosol models, different wavelengths and viewing geometries used in the retrievals, different parametrizations of ocean surface reflectance, etc. Comparisons of these satellite aerosol retrievals with the surface AERONET observations provide an opportunity to objectively evaluate as well as improve the accuracy of these satellite retrievals. Myhre et al. 2005b [SRC] ) showed that dedicated instruments using multi-channel and multi-view algorithms perform better when compared against AERONET than the simple algorithms that they have replaced, and Zhao et al. 2005 [JoC] ) showed that retrievals based on dynamic aerosol models perform better than those based on globally fixed aerosol models. While some systematic biases in specific satellite products exist (e.g., Jeong et al., 2005 [JoC] ; Remer et al., 2005 [JoC] ), these can be corrected for (e.g., Bellouin et al., 2005 [JoC, MoS, SRC] ; Kaufman et al., 2005b [MoS] ), which then enables an assessment of the direct radiative effect and the direct RF from an observational perspective, as detailed below.
2.4.2.1.2 Satellite retrievals of direct radiative effect
The solar direct radiative effect (DRE) is the sum of the direct effects due to anthropogenic and natural aerosol species while the direct RF only considers the anthropogenic components. Satellite estimates of the global clear-sky DRE over oceans have advanced since the TAR, owing to the development of dedicated aerosol instruments and algorithms, as summarised by Yu et al. 2006 [MoS] ) (see Table 2.3 ). Table 2.3 suggests a reasonable agreement of the global mean, diurnally averaged clear-sky DRE from various studies, with a mean of –5.4 Wm–2 and a standard deviation of 0.9 Wm–2.The clear-sky DRE is converted to an all-sky DRE by Loeb and Manalo-Smith 2005 [JoC] ) who estimated an all-sky DRE over oceans of –1.6 to –2.0 Wm–2 but assumed no aerosol contribution to the DRE from cloudy regions; such an assumption is not valid for optically thin clouds or if partially absorbing aerosols exist above the clouds (see Section 2.4.4.4 ).
Table 2.3 .The direct aerosol radiative effect (DRE) estimated from satellite remote sensing studies (adapted and updated from Yu et al., 2006 [MoS] ).
| Reference | Instrumenta | Data Analysed | Brief Description | Clear Sky DRE(Wm–2)ocean |
|---|---|---|---|---|
| Bellouin et al. (2005) t | MODIS; TOMS; SSM/I | 2002 | MODIS fine and total τaer with TOMS Aerosol Index and SSM/I todiscriminate dust from sea salt. | –6.8 |
| Loeb and Manalo-Smith (2005) | CERES; MODIS | Mar 2000 to Dec 2003 | CERES radiances/irradiances and angular distribution models and aerosol properties from either MODIS or fromNOAA-NESDISb algorithm used toestimate the direct radiative effect. | –3.8 (NESDIS)to –5.5 (MODIS) |
| Remer and Kaufman (2006) | MODIS | Aug 2001 to Dec 2003 | Best-prescribed aerosol model fitted to MODIS data. τaer from fine-mode fraction. | –5.7 ± 0.4 |
| Zhang et al. (2005); Christopher and Zhang (2004) | CERES; MODIS | Nov 2000 to Aug 2001 | MODIS aerosol properties, CERES radiances/irradiances and angulardistribution models used to estimate thedirect radiative effect. | –5.3 ± 1.7 |
| Bellouin et al. (2003) | POLDER | Nov 1996 to Jun 1997 | Best-prescribed aerosol model fitted to POLDER data | –5.2 |
| Loeb and Kato (2002) | CERES; VIRS | Jan 1998 to Aug 1998; Mar 2000. | τaer from VIRS regressed against the TOA CERES irradiance (35°N to 35°S) | –4.6 ± 1.0 |
| Chou et al. (2002) | SeaWiFs | 1998 | Radiative transfer calculations with SeaWiFS τaer and prescribed opticalproperties | –5.4 |
| Boucher and Tanré (2000) | POLDER | Nov 1996 to Jun 1997 | Best-prescribed aerosol model fitted to POLDER data | –5 to –6 |
| Haywood et al. (1999) | ERBE | Jul 1987 to Dec 1988 | DRE diagnosed from GCM-ERBE TOA irradiances | –6.7 |
| Mean (standard deviation) | –5.4 (0.9) |
Furthermore, use of a combination of sensors on the same satellite offers the possibility of concurrently deriving τaer and the DRE (e.g., Zhang and Christopher, 2003 [JoC, MoS] ; Zhang et al., 2005 [JoC, MoS] ), which enables estimation of the DRE efficiency, that is, the DRE divided by τaer (Wm–2 τaer–1). Because the DRE efficiency removes the dependence on the geographic distribution of τaer it is a useful parameter for comparison of models against observations (e.g., Anderson et al., 2005b [JoC, MoS] ); however, the DRE efficiency thus derived is not a linear function of τaer at high τaer such as those associated with intense mineral dust, biomass burning or pollution events.
2.4.2.1.3. Satellite retrievals of direct radiative forcing
Kaufman et al. 2005a [JoC, MoS] ) estimated the anthropogenic-only component of the aerosol fine-mode fraction from the MODIS product to deduce a clear sky RF over ocean of –1.4 Wm–2 .Christopher et al. 2006 [JoC, MoS] ) used a combination of the MODIS fine-mode fraction and Clouds and the Earth’s Radiant Energy System (CERES) broadband TOA fluxes and estimated an identical value of –1.4 ± 0.9 Wm–2 .Bellouin et al. 2005 [JoC, MoS, SRC] ) used a combination of MODIS τaer and fine-mode fraction together with data from AeroCom (see Section 2.4.3 )to determine an all-sky RF of aerosols over both land and ocean of –0.8 ± 0.2 Wm–2,but this does not include the contribution to the RF and associated uncertainty from cloudy skies. Chung et al. 2005 [JoC, MoS, SRC] ) performed a similar satellite/AERONET/model analysis, but included the contribution from cloudy areas to deduce an RF of –0.35 Wm–2 or –0.50 Wm–2 depending upon whether the anthropogenic fraction is determined from a model or from the MODIS fine-mode fraction and suggest an overall uncertainty range of –0.1 to –0.6 Wm–2 .Yu et al. 2006 [MoS] ) used several measurements to estimate a direct RF of –0.5 ± 0.33 Wm–2.These estimates of the RF are compared to those obtained from modelling studies in Section 2.4.4.7 .
2.4.2.2 Surface-Based Retrievals
Asignificant advancement since the TAR is the continued deployment and development of surface based remote sensing sun-photometer sites such as AERONET ( (Holben et al., 1998 ) ), and the establishment of networks of aerosol lidar systems such as the European Aerosol Research Lidar Network (EARLINET, Matthias et al., 2004 [JoC] ), the Asian Dust Network (ADNET, Murayama et al., 2001 [JoC] ), and the Micro-Pulse Lidar Network (MPLNET, Welton et al., 2001 [NPR] ).
The distribution of AERONET sites is also shown in Figure 2.11 (top panel). Currently there are approximately 150 sites operating at any one time, many of which are permanent to enable determination of climatological and interannual column-averaged monthly and seasonal means. In addition to measurements of τaer as a function of wavelength, new algorithms have been developed that measure sky radiance as a function of scattering angle ( Nakajima et al., 1996 [JoC, SRC] ; Dubovik and King, 2000 [JoC] ). From these measurements, the column-averaged size distribution and, if the τaer is high enough (τaer > 0.5), the aerosol single scattering albedo, ωo, and refractive indices may be determined at particular wavelengths ( Dubovik et al., 2000 [JoC] ), allowing partitioning between scattering and absorption. While these inversion products have not been comprehensively validated, a number of studies show encouraging agreement for both the derived size distribution and ωo when compared against in situ measurements by instrumented aircraft for different aerosol species (e.g., Dubovik et al., 2002 [JoC] ; Haywood et al., 2003a [JoC, SRC] ; Reid et al., 2003 [JoC] ; Osborne et al., 2004 [JoC, SRC] ). A climatology of the aerosol DRE based on the AERONET aerosols has also been derived ( Zhou et al., 2005 [JoC, MoS] ).
The MPLNET Lidar network currently consists of 11 lidars worldwide; 9 are co-located with AERONET sites and provide complementary vertical distributions of aerosol backscatter and extinction. Additional temporary MPLNET sites have supported major aerosol field campaigns (e.g., Campbell et al., 2003 [JoC] ). The European-wide lidar network EARLINET currently has 15 aerosol lidars making routine retrievals of vertical profiles of aerosol extinction (Mathias et al., 2004 ), and ADNET is a network of 12 lidars making routine measurements in Asia that have been used to assess the vertical profiles of Asian dust and pollution events (e.g., Husar et al., 2001 [JoC] ; Murayama et al., 2001 [JoC] ).
2.4.3 Advances in Modelling the Aerosol Direct Effect
Since the TAR, more complete aerosol modules in a larger number of global atmospheric models now provide estimates of the direct RF. Several models have resolutions better than 2° by 2° in the horizontal and more than 20 to 30 vertical levels; this represents a considerable enhancement over the models used in the TAR. Such models now include the most important anthropogenic and natural species. Tables 2.4 , 2.5 and 2.6 summarise studies published since the TAR. Some of the more complex models now account explicitly for the dynamics of the aerosol size distribution throughout the aerosol atmospheric lifetime and also parametrize the internal/external mixing of the various aerosol components in a more physically realistic way than in the TAR (e.g., Adams and Seinfeld, 2002 [JoC, MoS] ; Easter et al., 2004 [JoC, MoS] ; Stier et al., 2005 [MoS] ). Because the most important aerosol species are now included, a comparison of key model output parameters, such as the total τaer,against satellite retrievals and surface-based sun photometer and lidar observations is possible (see Sections 2.4.2 and 2.4.4 ). Progress with respect to modelling the indirect effects due to aerosol-cloud interactions is detailed in Section 2.4.5 and Section 7.5 .Several studies have explored the sensitivity of aerosol direct RF to current parametrization uncertainties. These are assessed in the following sections.
Major progress since the TAR has been made in the documentation of the diversity of current aerosol model simulations. Sixteen groups have participated in the Global Aerosol Model Intercomparison (AeroCom) initiative ( Kinne et al., 2006 [MoS, SRC] ). Extensive model outputs are available via a dedicated website ( Schulz et al., 2004 [NPR, MoS, SRC] ). Three model experiments (named A, B, and PRE) were analysed. Experiment A models simulate the years 1996, 1997, 2000 and 2001, or a five-year mean encompassing these years. The model emissions and parametrizations are those determined by each research group, but the models are driven by observed meteorological fields to allow detailed comparisons with observations, including those from MODIS, MISR and the AERONET sun photometer network. ExperimentB models use prescribed AeroCom aerosol emissions for the year 2000, and experiment PRE models use prescribed aerosol emissions for the year 1750 ( Dentener et al., 2006 [ARC] ; Schulz et al., 2006 [MoS, SRC] ). The model diagnostics included information on emission and deposition fluxes, vertical distribution and sizes, thus enabling a better understanding of the differences in lifetimes of the various aerosol components in the models.
This paragraph discusses AeroCom results from Textor et al. 2006 [MoS, SRC] ). The model comparison study found a wide range in several of the diagnostic parameters; these, in turn, indicate which aerosol parametrizations are poorly constrained and/or understood. For example, coarse aerosol fractions are responsible for a large range in the natural aerosol emission fluxes (dust: ±49% and sea salt: ±200%, where uncertainty is 1 standard deviation of inter-model range), and consequently in the dry deposition fluxes. The complex dependence of the source strength on wind speed adds to the problem of computing natural aerosol emissions. Dust emissions for the same time period can vary by a factor of two or more depending on details of the dust parametrization ( Luo et al., 2003 [JoC] ; Timmreck and Schulz, 2004 [JoC, MoS, SRC] ; Balkanski et al., 2004 [NPR] ; Zender, 2004 [JoC, MoS] ), and even depend on the reanalysis meteorological data set used ( Luo et al., 2003 [JoC] ). With respect to anthropogenic and natural emissions of other aerosol components, modelling groups tended to make use of similar best guess information, for example, recently revised emissions information available via the Global Emissions Inventory Activity (GEIA). The vertical aerosol distribution was shown to vary considerably, which is a consequence of important differences in removal and vertical mixing parametrizations. The inter-model range for the fraction of sulphate mass below 2.5 km to that of total sulphate is 45 ± 23%. Since humidification takes place mainly in the boundary layer, this source of inter-model variability increases the range of modelled direct RF. Additionally, differences in the parametrization of the wet deposition/vertical mixing process become more pronounced above 5 km altitude. Some models have a tendency to accumulate insoluble aerosol mass (dust and carbonaceous aerosols) at higher altitudes, while others have much more efficient wet removal schemes. Tropospheric residence times, defined here as the ratio of burden over sinks established for an equilibrated one-year simulation, vary by 20 to 30% for the fine-mode aerosol species. These variations are of interest, since they express the linearity of modelled emissions to aerosol burden and eventually to RF.
Considerable progress has been made in the systematic evaluation of global model results (see references in Tables 2.4 to 2.6 ). The simulated global τaer at a wavelength of 0.55 µm in models ranges from 0.11 to 0.14. The values compare favourably to those obtained by remote sensing from the ground (AERONET, about 0.135) and space (satellite composite, about 0.15) ( Kinne et al., 2003 [JoC, MoS, SRC] , 2006 ), but significant differences exist in regional and temporal distributions. Modelled absorption optical thickness has been suggested to be underestimated by a factor of two to four when compared to observations ( Sato et al., 2003 [JoC] ) and DRE efficiencies have been shown to be lower in models both for the global average and regionally ( Yu et al., 2006 [MoS] ) (see Section 2.4.4.7 ). A merging of modelled and observed fields of aerosol parameters through assimilation methods of different degrees of complexity has also been performed since the TAR (e.g., Yu et al., 2003 [JoC, MoS] ; Chung et al., 2005 [JoC, MoS, SRC] ). Model results are constrained to obtain present-day aerosol fields consistent with observations. Collins et al. 2001 [JoC, MoS, SRC] ) showed that assimilation of satellite-derived fields of τaer can reduce the model bias down to 10% with respect to daily mean τaer measured with a sun photometer at the Indian Ocean Experiment (INDOEX) station Kaashidhoo. Liu et al. 2005 [JoC, MoS] ) demonstrated similar efficient reduction of errors in τaer.The magnitude of the global dust cycle has been suggested to range between 1,500 and 2,600 Tg yr–1 by minimising the bias between model and multiple dust observations ( Cakmur et al., 2006 [NPR, JoC, MoS] Bates et al. 2006 [MoS] ) focused on three regions downwind of major urban/population centres and performed radiative transfer calculations constrained by intensive and extensive observational parameters to derive 24-hour average clear-sky DRE of –3.3 ± 0.47, –14 ± 2.6 and –6.4 ± 2.1 Wm–2 for the north Indian Ocean, the northwest Pacific and the northwest Atlantic, respectively. By constraining aerosol models with these observations, the uncertainty associated with the DRE was reduced by approximately a factor of two.
2.4.4 Estimates of Aerosol Direct Radiative Forcing
Unless otherwise stated, this section discusses the TOA direct RF of different aerosol types as a global annual mean quantity inclusive of the effects of clouds. Where possible, statistics from model results are used to assess the uncertainty in the RF. Recently published results and those grouped within AeroCom are assessed. Because the AeroCom results assessed here are based on prescribed emissions, the uncertainty in these results is lowered by having estimates of the uncertainties in the emissions. The quoted uncertainties therefore include the structural uncertainty (i.e., differences associated with the model formulation and structure) associated with the RF, but do not include the full range of parametric uncertainty (i.e., differences associated with the choice of key model parameters), as the model results are essentially best estimates constrained by observations of emissions, wet and dry deposition, size distributions, optical parameters, hygroscopicity, etc. ( Pan et al., 1997 [JoC, MoS, SRC] ). The uncertainties are reported as the 5 to 95% confidence interval to allow the uncertainty in the RF of each species of aerosol to be quantitatively intercompared.
2.4.4.1 Sulphate Aerosol
Atmospheric sulphate aerosol may be considered as consisting of sulphuric acid particles that are partly or totally neutralized by ammonia and that are present as liquid droplets or partly crystallized. Sulphate is formed by aqueous phase reactions within cloud droplets, oxidation of SO2 via gaseous phase reactions with OH, and by condensational growth onto pre-existing particles (e.g., Penner et al., 2001 [NPR, ARC] ). Emission estimates are summarised by Haywood and Boucher 2000 [JoC, MoS, SRC] ). The main source of sulphate aerosol is via SO2 emissions from fossil fuel burning (about 72%), with a small contribution from biomass burning (about 2%), while natural sources are from dimethyl sulphide emissions by marine phytoplankton (about 19%) and by SO2 emissions from volcanoes (about 7%). Estimates of global SO2 emissions range from 66.8 to 92.4 TgS yr–1 for anthropogenic emissions in the 1990 s and from 91.7 to 125.5 TgS yr–1 for total emissions. Emissions of SO2 from 25 countries in Europe were reduced from approximately 18 TgS yr–1 in 1980 to 4 TgS yr–1 in 2002 ( Vestreng et al., 2004 [NPR] ). In the USA, the emissions were reduced from about 12 to 8 TgS yr–1 in the period 1980 to 2000 ( EPA, 2003 [NPR] ). However, over the same period SO2 emissions have been increasing significantly from Asia, which is estimated to currently emit 17 TgS yr–1 ( Streets et al., 2003 [JoC] ), and from developing countries in other regions (e.g., Lefohn et al., 1999 [MoS] ; van Aardenne et al., 2001 [JoC] ; Boucher and Pham, 2002 [JoC, MoS, SRC] ). The most recent study ( (Stern, 2005 ) ) suggests a decrease in global anthropogenic emissions from approximately 73 to 54 TgS yr–1 over the period 1980 to 2000, with NH emission falling from 64 to 43 TgS yr–1 and SH emissions increasing from 9 to 11 TgS yr–1 .Smith et al. 2004 [NPR, ARC] ) suggested a more modest decrease in global emissions, by some 10 TgS yr–1 over the same period. The regional shift in the emissions of SO2 from the USA, Europe, Russia, Northern Atlantic Ocean and parts of Africa to Southeast Asia and the Indian and Pacific Ocean areas will lead to subsequent shifts in the pattern of the RF (e.g., Boucher and Pham, 2002 [JoC, MoS, SRC] ; Smith et al., 2004 [NPR, ARC] ; Pham et al., 2005 [JoC, MoS, SRC] ). The recently used emission scenarios take into account effective injection heights and their regional and seasonal variability (e.g., Dentener et al., 2006 [ARC] ).
The optical parameters of sulphate aerosol have been well documented (see Penner et al., 2001 [NPR, ARC] and references therein). Sulphate is essentially an entirely scattering aerosol across the solar spectrum (ωo = 1) but with a small degree of absorption in the near-infrared spectrum. Theoretical and experimental data are available on the relative humidity dependence of the specific extinction coefficient,fRH (e.g., ( Tang et al., 1995 ) ). Measurement campaigns concentrating on industrial pollution, such as the Tropospheric Aerosol Radiative Forcing Experiment (TARFOX; Russell et al., 1999 [JoC, MoS] ), the Aerosol Characterization Experiment (ACE-2; Raes et al., 2000 [JoC] ), INDOEX ( Ramanathan et al., 2001b [JoC, MoS, SRC] ), the Mediterranean Intensive Oxidants Study (MINOS, 2001 campaign), ACE-Asia ( 2001 ), Atmospheric Particulate Environment Change Studies (APEX, from 2000 to 2003 ), the New England Air Quality Study (NEAQS, in 2003 ) and the Chesapeake Lighthouse and Aircraft Measurements for Satellites (CLAMS; Smith et al., 2005 [JoC] ), continue to show that sulphate contributes a significant fraction of the sub-micron aerosol mass, anthropogenic τaer and RF (e.g., Hegg et al., 1997 [JoC] ; Russell and Heintzenberg, 2000 [JoC] ; Ramanathan et al., 2001b [JoC, MoS, SRC] ; Magi et al., 2005 [JoC] ; Quinn and Bates, 2005 [JoC] ). However, sulphate is invariably internally and externally mixed to varying degrees with other compounds such as biomass burning aerosol (e.g., Formenti et al., 2003 [JoC] ), fossil fuel black carbon (e.g., Russell and Heintzenberg, 2000 [JoC] ), organic carbon ( Novakov et al., 1997 [JoC] ; Brock et al., 2004 [JoC] ), mineral dust (e.g., Huebert et al., 2003 [JoC] ) and nitrate aerosol (e.g., Schaap et al., 2004 [MoS] ). This results in a composite aerosol state in terms of effective refractive indices, size distributions, physical state, morphology, hygroscopicity and optical properties.
The TAR reported an RF due to sulphate aerosol of –0.40 Wm–2 with an uncertainty of a factor of two, based on global modelling studies that were available at that time. Results from model studies since the TAR are summarised in Table 2.4 .For models A to L, the RF ranges from approximately –0.21 Wm–2 ( Takemura et al., 2005 [JoC, MoS, SRC] ) to –0.96 Wm–2 ( Adams et al., 2001 [JoC, MoS] ) with a mean of –0.46 Wm–2 and a standard deviation of 0.20 Wm–2.The range in the RF per unit τaer is substantial due to differing representations of aerosol mixing state, optical properties, cloud, surface reflectance, hygroscopic growth, sub-grid scale effects, radiative transfer codes, etc. ( Ramaswamy et al., 2001 [NPR, MoS, SRC] Myhre et al. 2004b [JoC, MoS, SRC] ) performed several sensitivity studies and found that the uncertainty was particularly linked to the hygroscopic growth and that differences in the model relative humidity fields could cause differences of up to 60% in the RF. The RFs from the models M to U participating in the AeroCom project are slightly weaker than those obtained from the other studies, with a mean of approximately –0.35 Wm–2 and a standard deviation of 0.15 Wm–2;the standard deviation is reduced for the AeroCom models owing to constraints on aerosol emissions, based on updated emission inventories (see Table 2.4 ). Including the uncertainty in the emissions reported in Haywood and Boucher 2000 [JoC, MoS, SRC] ) increases the standard deviation to 0.2 Wm–2.As sulphate aerosol is almost entirely scattering, the surface forcing will be similar or marginally stronger than the RF diagnosed at the TOA. The uncertainty in the RF estimate relative to the mean value remains relatively large compared to the situation for LLGHGs.
The mean and median of the sulphate direct RF from grouping all these studies together are identical at –0.41 Wm–2.Disregarding the strongest and weakest direct RF estimates to approximate the 90% confidence interval leads to an estimate of –0.4 ± 0.2 Wm–2.
Table 2.4 .The direct radiative forcing for sulphate aerosol derived from models published since the TAR and from the AeroCom simulations where different models used identical emissions. Load and aerosol optical depth (τaer )refer to the anthropogenic sulphate; τaer ant is the fraction of anthropogenic sulphate to total sulphate τaer for present day, NRFM is the normalised RF by mass, and NRF is the normalised RF per unit τaer .
| No | Modela | LOAD (mg(SO4)m–2) | τaer (0.55 µm) | τaer ant (%) | RF (Wm–2) | NRFM (Wg–1) | NRF (Wm–2 τaer–1) | Reference |
|---|---|---|---|---|---|---|---|---|
| Published since IPCC, 2001 | ||||||||
| A | CCM3 | 2.23 | –0.56 | –251 | (Kiehl et al., 2000) | |||
| B | GEOSCHEM | 1.53 | 0.018 | –0.33 | –216 | –18 | (Martin et al., 2004) | |
| C | GISS | 3.30 | 0.022 | –0.65 | –206 | –32 | (Koch, 2001) | |
| D | GISS | 3.27 | –0.96 | –293 | (Adams et al., 2001) | |||
| E | GISS | 2.12 | –0.57 | –269 | (Liao and Seinfeld, 2005) | |||
| F | SPRINTARS | 1.55 | 0.015 | 72 | –0.21 | –135 | –8 | (Takemura et al., 2005) |
| G | LMD | 2.76 | –0.42 | –152 | (Boucher and Pham., 2002) | |||
| H | LOA | 3.03 | 0.030 | –0.41 | –135 | –14 | (Reddy et al., 2005b) | |
| I | GATORG | 3.06 | –0.32 | –105 | (Jacobson, 2001a) | |||
| J | PNNL | 5.50 | 0.042 | –0.44 | –80 | –10 | (Ghan et al., 2001) | |
| K | UIO_CTM | 1.79 | 0.019 | –0.37 | –207 | –19 | (Myhre et al., 2004b) | |
| L | UIO_GCM | 2.28 | –0.29 | –127 | (Kirkevag and Iversen, 2002) | |||
| AeroCom: identical emissions used for year 1750 and 2000 | ||||||||
| M | UMI | 2.64 | 0.020 | 58 | –0.58 | –220 | –28 | (Liu and Penner, 2002) |
| N | UIO_CTM | 1.70 | 0.019 | 57 | –0.35 | –208 | –19 | (Myhre et al., 2003) |
| O | LOA | 3.64 | 0.035 | 64 | –0.49 | –136 | –14 | (Reddy and Boucher, 2004) |
| P | LSCE | 3.01 | 0.023 | 59 | –0.42 | –138 | –18 | (Schulz et al., 2006) |
| Q | ECHAM5-HAM | 2.47 | 0.016 | 60 | –0.46 | –186 | –29 | (Stier et al., 2005) |
| R | GISS | 1.34 | 0.006 | 41 | –0.19 | –139 | –31 | (Koch, 2001) |
| S | UIO_GCM | 1.72 | 0.012 | 59 | –0.25 | –145 | –21 | (Iversen and Seland, 2002; Kirkevag and Iversen, 2002) |
| T | SPRINTARS | 1.19 | 0.013 | 59 | –0.16 | –137 | –13 | (Takemura et al., 2005) |
| U | ULAQ | 1.62 | 0.020 | 42 | –0.22 | –136 | –11 | (Pitari et al., 2002) |
| Average A to L | 2.80 | 0.024 | –0.46 | –176 | –17 | |||
| Average M to U | 2.15 | 0.018 | 55 | –0.35 | –161 | –20 | ||
| Minimum A to U | 1.19 | 0.006 | 41 | –0.96 | –293 | –32 | ||
| Maximum A to U | 5.50 | 0.042 | 72 | –0.16 | –72 | –8 | ||
| Std. dev. A to L | 1.18 | 0.010 | 0.20 | 75 | 9 | |||
| Std. dev. M to U | 0.83 | 0.008 | 8 | 0.15 | 34 | 7 | ||
2.4.4.2 Organic Carbon Aerosol from Fossil Fuels
Organic aerosols are a complex mixture of chemical compounds containing carbon-carbon bonds produced from fossil fuel and biofuel burning and natural biogenic emissions. Organic aerosols are emitted as primary aerosol particles or formed as secondary aerosol particles from condensation of organic gases considered semi-volatile or having low volatility. Hundreds of different atmospheric organic compounds have been detected in the atmosphere (e.g., ( Hamilton et al., 2004; ) Murphy, 2005 [JoC] ), which makes definitive modelling of the direct and indirect effects extremely challenging ( McFiggans et al., 2006 [ARC] ). Emissions of primary organic carbon from fossil fuel burning have been estimated to be 10 to 30 TgC yr–1 ( Liousse et al., 1996 [JoC, MoS] ; Cooke et al., 1999 [JoC, MoS, ARC] ; Scholes and Andreae, 2000 [JoC] ). More recently, Bond et al. 2004 [JoC] ) provided a detailed analysis of primary organic carbon emissions from fossil fuels, biofuels and open burning, and suggested that contained burning (approximately the sum of fossil fuel and biofuel) emissions are in the range of 5 to 17 TgC yr–1,with fossil fuel contributing only 2.4 TgC yr–1 .Ito and Penner 2005 [JoC, ARC] ) estimated global fossil fuel particulate organic matter (POM, which is the sum of the organic carbon and the other associated chemical elements) emissions of around 2.2 Tg(POM) yr–1,and global biofuel emissions of around 7.5 Tg(POM) yr–1 .Ito and Penner 2005 [JoC, ARC] ) estimated that emissions of fossil and biofuel organic carbon increased by a factor of three over the period 1870 to 2000 . Subsequent to emission, the hygroscopic, chemical and optical properties of organic carbon particles continue to change because of chemical processing by gas-phase oxidants such as ozone, OH, and the nitrate radical (NO3)(e.g., Kanakidou et al., 2005 [MoS] ). Atmospheric concentrations of organic aerosol are frequently similar to those of industrial sulphate aerosol. Novakov et al. 1997 [JoC] and Hegg et al. 1997 [JoC] ) measured organic carbon in pollution off the East Coast of the USA during the TARFOX campaign, and found organic carbon primarily from fossil fuel burning contributed up to 40% of the total submicron aerosol mass and was frequently the most significant contributor to τaer.During INDOEX, which studied the industrial plume over the Indian Ocean, Ramanathan et al. 2001b [JoC, MoS, SRC] ) found that organic carbon was the second largest contributor to τaer after sulphate aerosol.
Observational evidence suggests that some organic aerosol compounds from fossil fuels are relatively weakly absorbing but do absorb solar radiation at some ultraviolet and visible wavelengths (e.g., ( Bond et al., 1999; ) Jacobson, 1999 [JoC] ; Bond, 2001 [JoC] ) although organic aerosol from high-temperature combustion such as fossil fuel burning ( Dubovik et al., 1998 [JoC] ; Kirchstetter et al., 2004 [JoC] ) appears less absorbing than from low-temperature combustion such as open biomass burning. Observations suggest that a considerable fraction of organic carbon is soluble to some degree, while at low relative humidity more water is often associated with the organic fraction than with inorganic material. At higher relative humidities, the hygroscopicity of organic carbon is considerably less than that of sulphate aerosol ( Kotchenruther and Hobbs, 1998 [JoC] ; Kotchenruther et al., 1999 [JoC] ).
Based on observations and fundamental chemical kinetic principles, attempts have been made to formulate organic carbon composition by functional group analysis in some main classes of organic chemical species (e.g., Decesari et al., 2000 [JoC, ARC] , 2001; ( Maria et al., 2002; ) ( Ming and Russell, 2002 ) ), capturing some general characteristics in terms of refractive indices, hygroscopicity and cloud activation properties. This facilitates improved parametrizations in global models (e.g., Fuzzi et al., 2001 [JoC, MoS, ARC] ; Kanakidou et al., 2005 [MoS] ; Ming et al., 2005a [JoC, MoS, SRC] ).
Organic carbon aerosol from fossil fuel sources is invariably internally and externally mixed to some degree with other combustion products such as sulphate and black carbon (e.g., Novakov et al., 1997 [JoC] ; Ramanathan et al., 2001b [JoC, MoS, SRC] ). Theoretically, coatings of essentially non-absorbing components such as organic carbon or sulphate on strongly absorbing core components such as black carbon can increase the absorption of the composite aerosol (e.g., Fuller et al., 1999 [JoC] ; Jacobson, 2001a [JoC, MoS] ; Stier et al., 2006a [JoC] ), with results backed up by laboratory studies (e.g., ( Schnaiter et al., 2003 ) ). However, coatings of organic carbon aerosol on hygroscopic aerosol such as sulphate may lead to suppression of the rate of water uptake during cloud activation ( (Xiong et al., 1998; ) Chuang, 2003 [JoC] ).
Current global models generally treat organic carbon using one or two tracers (e.g., water-insoluble tracer, water-soluble tracer) and highly parametrized schemes have been developed to represent the direct RF. Secondary organic carbon is highly simplified in the global models and in many cases treated as an additional source similar to primary organic carbon. Considerable uncertainties still exist in representing the refractive indices and the water of hydration associated with the particles because the aerosol properties will invariably differ depending on the combustion process, chemical processing in the atmosphere, mixing with the ambient aerosol, etc. (e.g., McFiggans et al., 2006 [ARC] ).
The TAR reported an RF of organic carbon aerosols from fossil fuel burning of –0.10 Wm–2 with a factor of three uncertainty. Many of the modelling studies performed since the TAR have investigated the RF of organic carbon aerosols from both fossil fuel and biomass burning aerosols, and the combined RF of both components. These studies are summarised in Table 2.5 .The RF from total organic carbon (POM) from both biomass burning and fossil fuel emissions from recently published models A to K and AeroCom models (L to T) is –0.24 Wm–2 with a standard deviation of 0.08 Wm–2 and –0.16 Wm–2 with a standard deviation of 0.10 Wm–2,respectively. Where the RF due to organic carbon from fossil fuels is not explicitly accounted for in the studies, an approximate scaling based on the source apportionment of 0.25:0.75 is applied for fossil fuel organic carbon:biomass burning organic carbon ( Bond et al., 2004 [JoC] ). The mean RF of the fossil fuel component of organic carbon from those studies other than in AeroCom is –0.06 Wm–2,while those from AeroCom produce an RF of –0.03 Wm–2 with a range of –0.01 Wm–2 to –0.06 Wm–2 and a standard deviation of around 0.02 Wm–2.Note that these RF estimates, to a large degree, only take into account primary emitted organic carbon. These studies all use optical properties for organic carbon that are either entirely scattering or only weakly absorbing and hence the surface forcing is only slightly stronger than that at the TOA.
The mean and median for the direct RF of fossil fuel organic carbon from grouping all these studies together are identical at –0.05 Wm–2 with a standard deviation of 0.03 Wm–2.The standard deviation is multiplied by 1.645 to approximate the 90% confidence interval. [9] This leads to a direct RF estimate of –0.05 ± 0.05 Wm–2.
2.4.4.3 Black Carbon Aerosol from Fossil Fuels
Black carbon (BC) is a primary aerosol emitted directly at the source from incomplete combustion processes such as fossil fuel and biomass burning and therefore much atmospheric BC is of anthropogenic origin. Global, present-day fossil fuel emission estimates range from 5.8 to 8.0 TgC yr–1 ( Haywood and Boucher, 2000 [JoC, MoS, SRC] and references therein). Bond et al. 2004 [JoC] ) estimated the total current global emission of BC to be approximately 8 TgC yr–1,with contributions of 4.6 TgC yr–1 from fossil fuel and biofuel combustion and 3.3 TgC yr–1 from open biomass burning, and estimated an uncertainty of about a factor of two. Ito and Penner 2005 [JoC, ARC] ) suggested fossil fuel BC emissions for 2000 of around 2.8 TgC yr–1.The trends in emission of fossil fuel BC have been investigated in industrial areas by Novakov et al. 2003 [JoC] and Ito and Penner 2005 [JoC, ARC] Novakov et al. 2003 [JoC] ) reported that significant decreases were recorded in the UK, Germany, the former Soviet Union and the USA over the period 1950 to 2000, while significant increases were reported in India and China. Globally, Novakov et al. 2003 [JoC] ) suggested that emissions of fossil fuel BC increased by a factor of three between 1950 and 1990 (2.2 to 6.7 TgC yr–1)owing to the rapid expansion of the USA, European and Asian economies (e.g., ( Streets et al., 2001, ) 2003 ), and have since fallen to around 5.6 TgC yr–1 owing to further emission controls. Ito and Penner 2005 [JoC, ARC] ) determined a similar trend in emissions over the period 1950 to 2000 of approximately a factor of three, but the absolute emissions are smaller than in Novakov et al. 2003 [JoC] ) by approximately a factor of 1.7.
Black carbon aerosol strongly absorbs solar radiation. Electron microscope images of BC particles show that they are emitted as complex chain structures (e.g., Posfai et al., 2003 [JoC] ), which tend to collapse as the particles age, thereby modifying the optical properties (e.g., Abel et al., 2003 [JoC] ). The Indian Ocean Experiment ( Ramanathan et al., 2001b [JoC, MoS, SRC] and references therein) focussed on emissions of aerosol from the Indian sub-continent, and showed the importance of absorption by aerosol in the atmospheric column. These observations showed that the local surface forcing (–23 Wm–2)was significantly stronger than the local RF at the TOA (–7 Wm–2). Additionally, the presence of BC in the atmosphere above highly reflective surfaces such as snow and ice, or clouds, may cause a significant positive RF ( Ramaswamy et al., 2001 [NPR, MoS, SRC] ). The vertical profile is therefore important, as BC aerosols or mixtures of aerosols containing a relatively large fraction of BC will exert a positive RF when located above clouds. Both microphysical (e.g., hydrophilic-to-hydrophobic nature of emissions into the atmosphere, aging of the aerosols, wet deposition) and meteorological aspects govern the horizontal and vertical distribution patterns of BC aerosols, and the residence time of these aerosols is thus sensitive to these factors ( Cooke et al., 2002 [JoC, MoS, SRC] ; Stier et al., 2006b [JoC, MoS] ).
The TAR assessed the RF due to fossil fuel BC as being +0.2 Wm–2 with an uncertainty of a factor of two. Those models since the TAR that explicitly model and separate out the RF due to BC from fossil fuels include those from Takemura et al. 2000 [JoC, MoS, SRC] Reddy et al. 2005a [JoC, SRC] and Hansen et al. 2005 [JoC, MoS, ARC] ) as summarised in Table 2.5 .The results from a number of studies that continue to group the RF from fossil fuel with that from biomass burning are also shown. Recently published results (A to K) and AeroCom studies (L to T) suggest a combined RF from both sources of +0.44 ± 0.13 Wm–2 and +0.29 ± 0.15 Wm–2 respectively. The stronger RF estimates from the models A to K appear to be primarily due to stronger sources and column loadings as the direct RF/column loading is similar at approximately 1.2 to 1.3 W mg–1 ( Table 2.5 ). Carbonaceous aerosol emission inventories suggest that approximately 34 to 38% of emissions come from biomass burning sources and the remainder from fossil fuel burning sources. Models that separate fossil fuel from biomass burning suggest an equal split in RF. This is applied to those estimates where the BC emissions are not explicitly separated into emission sources to provide an estimate of the RF due to fossil fuel BC. For the AeroCom results, the fossil fuel BC RF ranges from +0.08 to +0.18 Wm–2 with a mean of +0.13 Wm–2 and a standard deviation of 0.03 Wm–2.For model results A to K, the RFs range from +0.15 Wm–2 to approximately +0.27 Wm–2,with a mean of +0.25 Wm–2 and a standard deviation of 0.11 Wm–2.
The mean and median of the direct RF for fossil fuel BC from grouping all these studies together are +0.19 and +0.16 Wm–2,respectively, with a standard deviation of nearly 0.10 Wm–2.The standard deviation is multiplied by 1.645 to approximate the 90% confidence interval and the best estimate is rounded upwards slightly for simplicity, leading to a direct RF estimate of +0.20 ± 0.15 W m–2. This estimate does not include the semi-direct effect or the BC impact on snow and ice surface albedo (see Sections 2.5.4 and 2.8.5.6 )
2.4.4.4 Biomass Burning Aerosol
The TAR reported a contribution to the RF of roughly –0.4 Wm–2 from the scattering components (mainly organic carbon and inorganic compounds) and +0.2 Wm–2 from the absorbing components (BC) leading to an estimate of the RF of biomass burning aerosols of –0.20 Wm–2 with a factor of three uncertainty. Note that the estimates of the BC RF from Hansen and Sato 2001 [JoC, MoS, ARC] Hansen et al. 2002 [JoC, MoS, ARC] Hansen and Nazarenko 2004 [JoC, MoS, ARC] and Jacobson 2001a [JoC, MoS] ) include the RF component of BC from biomass burning aerosol. Radiative forcing due to biomass burning (primarily organic carbon, BC and inorganic compounds such as nitrate and sulphate) is grouped into a single RF, because biomass burning emissions are essentially uncontrolled. Emission inventories show more significant differences for biomass burning aerosols than for aerosols of fossil fuel origin ( Kasischke and Penner, 2004 [JoC, MoS, ARC] ). Furthermore, the pre-industrial levels of biomass burning aerosols are also difficult to quantify ( Ito and Penner, 2005 [JoC, ARC] ; Mouillot et al., 2006 [JoC] ).
The Southern African Regional Science Initiative (SAFARI 2000 : see ( Swap et al., 2002, ) 2003 ) took place in 2000 and 2001 . The main objectives of the aerosol research were to investigate pyrogenic and biogenic emissions of aerosol in southern Africa ( Eatough et al., 2003 [JoC] ; Formenti et al., 2003 [JoC] ; Hély et al., 2003 [JoC] ), validate the remote sensing retrievals ( Haywood et al., 2003b [JoC, SRC] ; Ichoku et al., 2003 [JoC, MoS] ) and to study the influence of aerosols on the radiation budget via the direct and indirect effects (e.g., Bergstrom et al., 2003 [JoC, MoS] ; Keil and Haywood, 2003 [JoC, MoS, SRC] ; Myhre et al., 2003 [JoC, MoS, SRC] ; Ross et al., 2003 [JoC] ). The physical and optical properties of fresh and aged biomass burning aerosol were characterised by making intensive observations of aerosol size distributions, optical properties, and DRE through in situ aircraft measurements (e.g., Abel et al., 2003 [JoC] ; Formenti et al., 2003 [JoC] ; Haywood et al., 2003b [JoC, SRC] ; Magi and Hobbs, 2003 [JoC] ; Kirchstetter et al., 2004 [JoC] ) and radiometric measurements (e.g., Bergstrom et al., 2003 [JoC, MoS] ; Eck et al., 2003 [JoC, MoS] ). The ωo at 0.55 µm derived from near-source AERONET sites ranged from 0.85 to 0.89 ( Eck et al., 2003 [JoC, MoS] ), while ωo at 0.55 µm for aged aerosol was less absorbing at approximately 0.91 ( Haywood et al., 2003b [JoC, SRC] Abel et al. 2003 [JoC] ) showed evidence that ωo at 0.55 µm increased from approximately 0.85 to 0.90 over a time period of approximately two hours subsequent to emission, and attributed the result to the condensation of essentially non-absorbing organic gases onto existing aerosol particles. Fresh biomass burning aerosols produced by boreal forest fires appear to have weaker absorption than those from tropical fires, with ωo at 0.55 µm greater than 0.9 ( Wong and Li 2002 [JoC] ). Boreal fires may not exert a significant direct RF because a large proportion of the fires are of natural origin and no significant change over the industrial era is expected. However, Westerling et al. 2006 [JoC, ARC] ) showed that earlier spring and higher temperatures in USA have increased wildfire activity and duration. The partially absorbing nature of biomass burning aerosol means it exerts an RF that is larger at the surface and in the atmospheric column than at the TOA (see Figure 2.12 ).
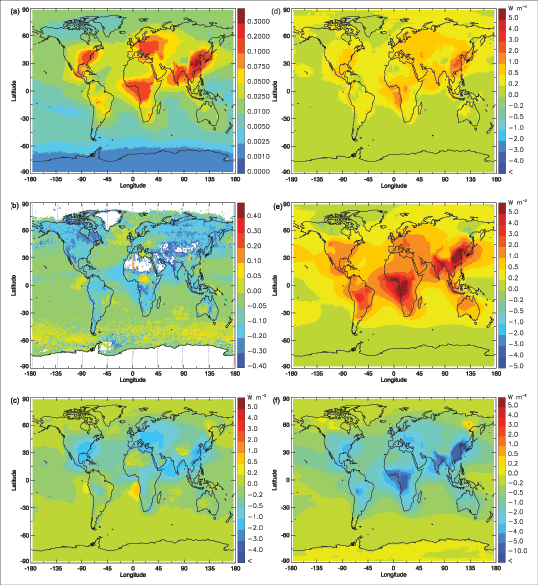
Figure 2.12. Characteristic aerosol properties related to their radiative effects, derived as the mean of the results from the nine AeroCom models listed in Table 2.5 .All panels except (b) relate to the combined anthropogenic aerosol effect. Panel (b) considers the total (natural plus anthropogenic) aerosol optical depth from the models. (a) Aerosol optical depth. (b) Difference in total aerosol optical depth between model and MODIS data. (c) Shortwave RF. (d) Standard deviation of RF from the model results. (e) Shortwave forcing of the atmosphere. (f) Shortwave surface forcing.
Modelling efforts have used data from measurement campaigns to improve the representation of the physical and optical properties as well as the vertical profile of biomass burning aerosol ( Myhre et al., 2003 [JoC, MoS, SRC] ; Penner et al., 2003 [JoC, ARC] ; Section 2.4.5 ). These modifications have had important consequences for estimates of the RF due to biomass burning aerosols because the RF is significantly more positive when biomass burning aerosol overlies cloud than previously estimated ( Keil and Haywood, 2003 [JoC, MoS, SRC] ; Myhre et al., 2003 [JoC, MoS, SRC] ; Abel et al., 2005 [SRC] ). While the RF due to biomass burning aerosol in clear skies is certainly negative, the overall RF of biomass burning aerosol may be positive. In addition to modelling studies, observations of this effect have been made with satellite instruments. Hsu et al. 2003 [JoC] ) used SeaWiFs, TOMS and CERES data to show that biomass burning aerosol emitted from Southeast Asia is frequently lifted above the clouds, leading to a reduction in reflected solar radiation over cloudy areas by up to 100 Wm–2,and pointed out that this effect could be due to a combination of direct and indirect effects. Similarly, Haywood et al. 2003a [JoC, SRC] ) showed that remotely sensed cloud liquid water and effective radius underlying biomass burning aerosol off the coast of Africa are subject to potentially large systematic biases. This may have important consequences for studies that use correlations of τaer and cloud effective radius in estimating the indirect radiative effect of aerosols.
Since the biomass burning aerosols can exert a significant positive RF when above clouds, the aerosol vertical profile is critical in assessing the magnitude and even the sign of the direct RF in cloudy areas. Textor et al. 2006 [MoS, SRC] ) showed that there are significant differences in aerosol vertical profiles between global aerosol models. These differences are evident in the results from the recently published studies and AeroCom models in Table 2.5 .The most negative RF of –0.05 Wm–2 is from the model of Koch 2001 [JoC, MoS] ) and from the Myhre et al. 2003 [JoC, MoS, SRC] ) AeroCom submission, while several models have RFs that are slightly positive. Hence, even the sign of the RF due to biomass burning aerosols is in question.
Table 2.5 .Estimates of anthropogenic carbonaceous aerosol forcing derived from models published since the TAR and from the AeroCom simulations where different models used identical emissions. POM: particulate organic matter; BC: black carbon; BCPOM: BC and POM; FFBC: fossil fuel black carbon; FFPOM: fossil fuel particulate organic matter; BB: biomass burning sources included.
| No | Modela | LOAD POM (mgPOMm–2) | τaer POM | τaer POMant (%) | LOAD BC (mgm–2) | RF BCPOM (Wm–2) | RF POM (Wm–2) | RF BC (Wm–2) | RF FFPOM (Wm–2) | RF FFBC (Wm–2) | RF BB (Wm–2) | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Published since IPCC, 2001 | ||||||||||||
| A | SPRINT | 0.12 | –0.24 | 0.36 | –0.05 | 0.15 | –0.01 | (Takemura et al., 2001) | ||||
| B | LOA | 2.33 | 0.016 | 0.37 | 0.30 | –0.25 | 0.55 | –0.02 | 0.19 | 0.14 | (Reddy et al., 2005b) | |
| C | GISS | 1.86 | 0.017 | 0.29 | 0.35 | –0.26 | 0.61 | –0.13 | 0.49 | 0.065 | (Hansen et al., 2005) | |
| D | GISS | 1.86 | 0.015 | 0.29 | 0.05 | –0.30 | 0.35 | –0.08b | 0.18b | –0.05b | (Koch, 2001) | |
| E | GISS | 2.39 | 0.39 | 0.32 | –0.18 | 0.50 | –0.05b | 0.25b | 0.12b | (Chung and Seinfeld., 2002) | ||
| F | GISS | 2.49 | 0.43 | 0.30 | –0.23 | 0.53 | –0.06b | 0.27b | 0.09b | (Liao and Seinfeld, 2005) | ||
| G | SPRINTARS | 2.67 | 0.029 | 82 | 0.53 | 0.15 | –0.27 | 0.42 | –0.07b | 0.21b | 0.01b | (Takemura et al., 2005) |
| H | GATORG | 2.55 | 0.39 | 0.47 | –0.06 | 0.55 | –0.01b | 0.27b | 0.22b | (Jacobson, 2001b) | ||
| I | MOZGN | 3.03 | 0.018 | –0.34 | (Ming et al., 2005a) | |||||||
| J | CCM | 0.33 | 0.34 | (Wang, 2004) | ||||||||
| K | UIO-GCM | 0.30 | 0.19 | (Kirkevag and Iversen, 2002) | ||||||||
| AeroCom: identical emissions used for year 1750 and 2000 (Schulz et al., 2006) | ||||||||||||
| L | UMI | 1.16 | 0.0060 | 53 | 0.19 | 0.02 | –0.23 | 0.25 | –0.06b | 0.12b | –0.01 | (Liu and Penner, 2002) |
| M | UIO_CTM | 1.12 | 0.0058 | 55 | 0.19 | 0.02 | –0.16b | 0.22b | –0.04 | 0.11 | –0.05 | (Myhre et al., 2003) |
| N | LOA | 1.41 | 0.0085 | 52 | 0.25 | 0.14 | –0.16c | 0.32c | –0.04b | 0.16b | 0.02b | (Reddy and Boucher, 2004) |
| O | LSCE | 1.50 | 0.0079 | 46 | 0.25 | 0.13 | –0.17 | 0.30 | –0.04b | 0.15b | 0.02b | (Schulz et al., 2006) |
| P | ECHAM5-HAM | 1.00 | 0.0077 | 0.16 | 0.09 | –0.10c | 0.20c | –0.03b | 0.10b | 0.01 | (Stier et al., 2005) | |
| Q | GISS | 1.22 | 0.0060 | 51 | 0.24 | 0.08 | –0.14 | 0.22 | –0.03b | 0.11b | 0.01b | (Koch, 2001) |
| R | UIO_GCM | 0.88 | 0.0046 | 59 | 0.19 | 0.24 | –0.06 | 0.36 | –0.02b | 0.18b | 0.08b | (Iversen and Seland, 2002) |
| S | SPRINTARS | 1.84 | 0.0200 | 49 | 0.37 | 0.22 | –0.10 | 0.32 | –0.01 | 0.13 | 0.06 | (Takemura et al., 2005) |
| T | ULAQ | 1.71 | 0.0075 | 58 | 0.38 | –0.01 | –0.09 | 0.08 | –0.02b | 0.04b | –0.03b | (Pitari et al., 2002) |
| Average A–K | 2.38 | 0.019 | 0.38 | 0.26 | –0.24 | 0.44 | –0.06 | 0.25 | 0.07 | |||
| Average L–T | 1.32 | 0.008 | 53 | 0.25 | 0.10 | –0.13 | 0.25 | –0.03 | 0.12 | 0.01 | ||
| Stddev A–K | 0.42 | 0.006 | 0.08 | 0.14 | 0.08 | 0.13 | 0.04 | 0.11 | 0.09 | |||
| Stddev L–T | 0.32 | 0.005 | 4 | 0.08 | 0.09 | 0.05 | 0.08 | 0.01 | 0.04 | 0.04 | ||
The mean and median of the direct RF for biomass burning aerosol from grouping all these studies together are similar at +0.04 and +0.02 Wm–2,respectively, with a standard deviation of 0.07 Wm–2.The standard deviation is multiplied by 1.645 to approximate the 90% confidence interval, leading to a direct RF estimate of +0.03 ± 0.12 Wm–2.This estimate of the direct RF is more positive than that of the TAR owing to improvements in the models in representing the absorption properties of the aerosol and the effects of biomass burning aerosol overlying clouds.
2.4.4.5 Nitrate Aerosol
Atmospheric ammonium nitrate aerosol forms if sulphate aerosol is fully neutralised and there is excess ammonia. The direct RF due to nitrate aerosol is therefore sensitive to atmospheric concentrations of ammonia as well as NOx emissions. In addition, the weakening of the RF of sulphate aerosol in many regions due to reduced emissions( Section 2.4.4.1 )will be partially balanced by increases in the RF of nitrate aerosol (e.g., Liao and Seinfeld, 2005 [JoC, MoS] ). The TAR did not quantify the RF due to nitrate aerosol owing to the large discrepancies in the studies available at that time. van Dorland 1997 [JoC, MoS, SRC] and Jacobson 2001a [JoC, MoS] ) suggested relatively minor global mean RFs of –0.03 and –0.05 Wm–2,respectively, while Adams et al. 2001 [JoC, MoS] ) suggested a global mean RF as strong as –0.22 Wm–2.Subsequent studies include those of Schaap et al. 2004 [MoS] ), who estimated that the RF of nitrate over Europe is about 25% of that due to sulphate aerosol, and of Martin et al. 2004 [MoS] ), who reported –0.04 to –0.08 Wm–2 for global mean RF due to nitrate. Further, Liao and Seinfeld 2005 [JoC, MoS] ) estimated a global mean RF due to nitrate of –0.16 Wm–2.In this study, heterogeneous chemistry reactions on particles were included; this strengthens the RF due to nitrate and accounts for 25% of its RF. Feng and Penner 2007 [JoC, MoS, ARC, 2007] ) estimated a large, global, fine-mode nitrate burden of 0.58 mg NO3 m–2,which would imply an equivalent of 20% of the mean anthropogenic sulphate burden. Surface observations of fine-mode nitrate particles show that high concentrations are mainly found in highly industrialised regions, while low concentrations are found in rural areas ( Malm et al., 2004 [JoC] ;( Putaud et al., 2004 ) ). Atmospheric nitrate is essentially non-absorbing in the visible spectrum, and laboratory studies have been performed to determine the hygroscopicity of the aerosols (e.g., Tang 1997 [JoC] ; Martin et al., 2004 [MoS] and references therein). In the AeroCom exercise, nitrate aerosols were not included so fewer estimates of this compound exist compared to the other aerosol species considered.
The mean direct RF for nitrate is estimated to be –0.10 Wm–2 at the TOA, and the conservative scattering nature means a similar flux change at the surface. However, the uncertainty in this estimate is necessarily large owing to the relatively small number of studies that have been performed and the considerable uncertainty in estimates, for example, of the nitrate τaer.Thus, a direct RF of –0.10 ± 0.10 Wm–2 is tentatively adopted, but it is acknowledged that the number of studies performed is insufficient for accurate characterisation of the magnitude and uncertainty of the RF.
2.4.4.6 Mineral Dust Aerosol
Mineral dust from anthropogenic sources originates mainly from agricultural practices (harvesting, ploughing, overgrazing), changes in surface water (e.g., Caspian and Aral Sea, Owens Lake) and industrial practices (e.g., cement production, transport) ( Prospero et al., 2002 [JoC] ). The TAR reported that the RF due to anthropogenic mineral dust lies in the range of +0.4 to –0.6 Wm–2,and did not assign a best estimate because of the difficulties in determining the anthropogenic contribution to the total dust loading and the uncertainties in the optical properties of dust and in evaluating the competing shortwave and longwave radiative effects. For the sign and magnitude of the mineral dust RF, the most important factor for the shortwave RF is the single scattering albedo whereas the longwave RF is dependent on the vertical profile of the dust.
Tegen and Fung 1995 [JoC, ARC] ) estimated the anthropogenic contribution to mineral dust to be 30 to 50% of the total dust burden in the atmosphere. Tegen et al. 2004 [JoC, MoS] ) provided an updated, alternative estimate by comparing observations of visibility, as a proxy for dust events, from over 2,000 surface stations with model results, and suggested that only 5 to 7% of mineral dust comes from anthropogenic agricultural sources. Yoshioka et al. 2005 [JoC, MoS, ARC] ) suggested that a model simulation best reproduces the North African TOMS aerosol index observations when the cultivation source in the Sahel region contributes 0 to 15% to the total dust emissions in North Africa. A 35-year dust record established from Barbados surface dust and satellite observations from TOMS and the European geostationary meteorological satellite (Meteosat) show the importance of climate control and Sahel drought for interannual and decadal dust variability, with no overall trend yet documented ( Chiapello et al., 2005 [JoC] ). As further detailed in Section 7.3 ,climate change and CO2 variations on various time scales can change vegetation cover in semi-arid regions. Such processes dominate over land use changes as defined above, which would give rise to anthropogenic dust emissions ( Mahowald and Luo, 2003 [JoC, MoS] ; Moulin and Chiapello, 2004 [JoC] ; Tegen et al., 2004 [JoC, MoS] ). A best guess of 0 to 20% anthropogenic dust burden from these works is used here, but it is acknowledged that a very large uncertainty remains because the methods used cannot exclude either a reduction of 24% in present-day dust nor a large anthropogenic contribution of up to 50% ( Mahowald and Luo, 2003 [JoC, MoS] ; Mahowald et al., 2004 [JoC, MoS] ; Tegen et al., 2005 [NPR, JoC, MoS] ). The RF efficiency of anthropogenic dust has not been well differentiated from that of natural dust and it is assumed that they are equal. The RF due to dust emission changes induced by circulation changes between 1750 and the present are difficult to quantify and not included here (see also Section 7.5 ).
In situ measurements of the optical properties of local Saharan dust (e.g., Haywood et al., 2003c [JoC, SRC] ; Tanré et al., 2003 [JoC, MoS] ), transported Saharan mineral dust (e.g., Kaufman et al., 2001 [JoC] ; Moulin et al., 2001 [JoC] ;( Coen et al., 2004 ) ) and Asian mineral dust ( Huebert et al., 2003 [JoC] ; Clarke et al., 2004 [JoC] ; Shi et al., 2005 [MoS] ; Mikami et al., 2006 [JoC] ) reveal that dust is considerably less absorbing in the solar spectrum than suggested by previous dust models such as that of WMO 1986 [NPR] ). These new, spectral, simultaneous remote and in situ observations suggest that the single scattering albedo (ωo)of pure dust at a wavelength of 0.67 µm is predominantly in the range 0.90 to 0.99, with a central global estimate of 0.96. This is in accordance with the bottom-up modelling of ωo based on the haematite content in desert dust sources ( Claquin et al., 1999 [JoC, MoS, SRC] ; Shi et al., 2005 [MoS] ). Analyses of ωo from long-term AERONET sites influenced by Saharan dust suggest an average ωo of 0.95 at 0.67 µm ( Dubovik et al., 2002 [JoC] ), while unpolluted Asian dust during the Aeolian Dust Experiment on Climate (ADEC) had an average ωo of 0.93 at 0.67 µm ( Mikami et al., 2006 [JoC] and references therein). These high ωo values suggest that a positive RF by dust in the solar region of the spectrum is unlikely. However, absorption by particles from source regions with variable mineralogical distributions is generally not represented by global models.
Measurements of the DRE of mineral dust over ocean regions, where natural and anthropogenic contributions are indistinguishably mixed, suggest that the local DRE may be extremely strong: Haywood et al. 2003b [JoC, SRC] ) made aircraft-based measurements of the local instantaneous shortwave DRE of as strong as –130 Wm–2 off the coast of West Africa. Hsu et al. 2000 [JoC, MoS] ) used Earth Radiation Budget Experiment (ERBE) and TOMS data to determine a peak monthly mean shortwave DRE of around –45 Wm–2 for July 1985 . Interferometer measurements from aircraft and the surface have now measured the spectral signature of mineral dust for a number of cases (e.g., Highwood et al., 2003 [JoC] ) indicating an absorption peak in the centre of the 8 to 13 µm atmospheric window. Hsu et al. 2000 [JoC, MoS] ) determined a longwave DRE over land areas of North Africa of up to +25 Wm–2 for July 1985; similar results were presented by Haywood et al. 2005 [JoC, SRC] ) who determined a peak longwave DRE of up to +50 Wm–2 at the top of the atmosphere for July 2003 .
Recent model simulations report the total anthropogenic and natural dust DRE, its components and the net effect as follows (shortwave / longwave = net TOA, in Wm–2): H. Liao et al. 2004 [Ambiguous] ): –0.21 / +0.31 = +0.1; Reddy et al. 2005a [JoC, SRC] ): –0.28 / +0.14 = –0.14; Jacobson 2001a [JoC, MoS] ): –0.20 / +0.07 = –0.13; reference case and [range] of sensitivity experiments in Myhre and Stordal 2001a [JoC, MoS, SRC] , except case 6 and 7): –0.53 [–1.4 to +0.2] / +0.13 [+0.0 to +0.8] = –0.4 [–1.4 to +1.0]; and from AeroCom database models, GISS: –0.75 / (+0.19) = (–0.56); UIO-CTM*: –0.56 / (+0.19) = (–0.37); LSCE*: –0.6 / +0.3 = –0.3; UMI*: –0.54 / (+0.19) = (–0.35). (See Table 2.4 ,Note (a) for model descriptions.) The (*) star marked models use a single scattering albedo (approximately 0.96 at 0.67 µm) that is more representative of recent measurements and show more negative shortwave effects. A mean longwave DRE of 0.19 Wm–2 is assumed for GISS, UMI and UIO-CTM. The scatter of dust DRE estimates reflects the fact that dust burden and τaer vary by ±40 and ±44%, respectively, computed as standard deviation from 16 AeroCom A model simulations ( Textor et al., 2006 [MoS, SRC] ; Kinne et al., 2006 [MoS, SRC] ). Dust emissions from different studies range between 1,000 and 2,150 Tg yr–1 ( Zender, 2004 [JoC, MoS] ). Finally, a major effect of dust may be in reducing the burden of anthropogenic species at sub-micron sizes and reducing their residence time ( Bauer and Koch, 2005 [JoC, MoS] ; see Section 2.4.5.7).
The range of the reported dust net DRE (–0.56 to +0.1 Wm–2), the revised anthropogenic contribution to dust DRE of 0 to 20% and the revised absorption properties of dust support a small negative value for the anthropogenic direct RF for dust of –0.1 Wm–2.The 90% confidence level is estimated to be ±0.2 Wm–2,reflecting the uncertainty in total dust emissions and burdens and the range of possible anthropogenic dust fractions. At the limits of this uncertainty range, anthropogenic dust RF is as negative as –0.3 Wm–2 and as positive as +0.1 Wm–2.This range includes all dust DREs reported above, assuming a maximum 20% anthropogenic dust fraction, except the most positive DRE from Myhre and Stordal 2001a [JoC, MoS, SRC] ).
2.4.4.7 Direct RF for Combined Total Aerosol
The TAR reported RF values associated with several aerosol components but did not provide an estimate of the overall aerosol RF. Improved and intensified in situ observations and remote sensing of aerosols suggest that the range of combined aerosol RF is now better constrained. For model results, extensive validation now exists for combined aerosol properties, representing the whole vertical column of the atmosphere, such as τaer.Using a combined estimate implicitly provides an alternative procedure to estimating the RF uncertainty. This approach may be more robust than propagating uncertainties from all individual aerosol components. Furthermore, a combined RF estimate accounts for nonlinear processes due to aerosol dynamics and interactions between radiation field and aerosols. The role of nonlinear processes of aerosol dynamics in RF has been recently studied in global aerosol models that account for the internally mixed nature of aerosol particles ( Jacobson, 2001a [JoC, MoS] ; Kirkevåg and Iversen, 2002 [JoC, MoS] ; Liao and Seinfeld, 2005 [JoC, MoS] ; Takemura et al., 2005 [JoC, MoS, SRC] ; Stier et al., 2006b [JoC, MoS] ). Mixing of aerosol particle populations influences the radiative properties of the combined aerosol, because mixing changes size, chemical composition, state and shape, and this feed backs to the aerosol removal and formation processes itself. Chung and Seinfeld 2002 [JoC, MoS] ), in reviewing studies where BC is mixed either externally or internally with various other components, showed that BC exerts a stronger positive direct RF when mixed internally. Although the source-related processes for anthropogenic aerosols favour their submicron nature, natural aerosols enter the picture by providing a condensation surface for aerosol precursor gases. Heterogeneous reactions on sea salt and dust can reduce the sub-micron sulphate load by 28% (H. Liao et al., 2004 [Ambiguous] ) thereby reducing the direct and indirect RFs. Bauer and Koch 2005 [JoC, MoS] ) estimated the sulphate RF to weaken from –0.25 to –0.18 Wm–2 when dust is allowed to interact with the sulphur cycle. It would be useful to identify the RF contribution attributable to different source categories( Section 2.9.3 investigates this). However, few models have separated out the RF from specific emission source categories. Estimating the combined aerosol RF is a first step to quantify the anthropogenic perturbation to the aerosol and climate system caused by individual source categories.
Acentral model-derived estimate for the aerosol direct RF is based here on a compilation of recent simulation results using multi-component global aerosol models (see Table 2.6 ). This is a robust method for several reasons. The complexity of multi-component aerosol simulations captures nonlinear effects. Combining model results removes part of the errors in individual model formulations. As shown by Textor et al. 2006 [MoS, SRC] ), the model-specific treatment of transport and removal processes is partly responsible for the correlated dispersion of the different aerosol components. A less dispersive model with smaller burdens necessarily has fewer scattering and absorbing aerosols interacting with the radiation field. An error in accounting for cloud cover would affect the all-sky RF from all aerosol components. Such errors result in correlated RF efficiencies for major aerosol components within a given model. Directly combining aerosol RF results gives a more realistic aerosol RF uncertainty estimate. The AeroCom compilation suggests significant differences in the modelled local and regional composition of the aerosol (see also Figure 2.12 ), but an overall reproduction of the total τaer variability can be performed ( Kinne et al., 2006 [MoS, SRC] ). The scatter in model performance suggests that currently no preference or weighting of individual model results can be used ( Kinne et al., 2006 [MoS, SRC] ). The aerosol RF taken together from several models is more robust than an analysis per component or by just one model. The mean estimate from Table 2.6 of the total aerosol direct RF is –0.2 Wm–2,with a standard deviation of ±0.2 Wm–2.This is a low-end estimate for both the aerosol RF and uncertainty because nitrate (estimated as –0.1 Wm–2,see Section 2.4.4.5 )and anthropogenic mineral dust (estimated as –0.1 Wm–2,see Section 2.4.4.6 )are missing in most of the model simulations. Adding their contribution yields an overall model-derived aerosol direct RF of –0.4 Wm–2 (90% confidence interval: 0 to –0.8 Wm–2).
Three satellite-based measurement estimates of the aerosol direct RF have become available, which all suggest a more negative aerosol RF than the model studies (see Section 2.4.2.1.3). Bellouin et al. 2005 [JoC, MoS, SRC] ) computed a TOA aerosol RF of –0.8 ± 0.1 Wm–2 .Chung et al. 2005 [JoC, MoS, SRC] ), based upon similarly extensive calculations, estimated the value to be –0.35 ± 0.25 Wm–2, and Yu et al. 2006 [MoS] ) estimated it to be –0.5 ± 0.33 Wm–2.A central measurement-based estimate would suggest an aerosol direct RF of –0.55 Wm–2. Figure 2.13 shows the observationally based aerosol direct RF estimates together with the model estimates published since the TAR.
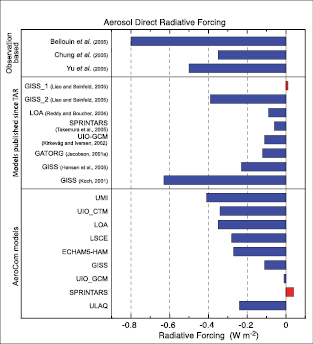
Figure 2.13. Estimates of the direct aerosol RF from observationally based studies, independent modelling studies, and AeroCom results with identical aerosol and aerosol precursor emissions. GISS_1 refers to a study employing an internal mixture of aerosol, and GISS_2 to a study employing an external mixture. See Table 2.4 ,Note (a) for descriptions of models.
The discrepancy between measurements and models is also apparent in oceanic clear-sky conditions where the measurement-based estimate of the combined aerosol DRE including natural aerosols is considered unbiased. In these areas, models underestimate the negative aerosol DRE by 20 to 40% ( Yu et al., 2006 [MoS] ). The anthropogenic fraction of τaer is similar between model and measurement based studies. Kaufman et al. 2005a [JoC, MoS] ) used satellite-observed fine-mode τaer to estimate the anthropogenic τaer.Correcting for fine-mode τaer contributions from dust and sea salt, they found 21% of the total τaer to be anthropogenic, while Table 2.6 suggests that 29% of τaer is anthropogenic. Finally, cloud contamination of satellite products, aerosol absorption above clouds, not accounted for in some of the measurement-based estimates, and the complex assumptions about aerosol properties in both methods can contribute to the present discrepancy and increase uncertainty in aerosol RF.
Alarge source of uncertainty in the aerosol RF estimates is associated with aerosol absorption. Sato et al. 2003 [JoC] ) determined the absorption τaer from AERONET measurements and suggested that aerosol absorption simulated by global aerosol models is underestimated by a factor of two to four. Schuster et al. 2005 [JoC] ) estimated the BC loading over continental-scale regions. The results suggest that the model concentrations and absorption τaer of BC from models are lower than those derived from AERONET. Some of this difference in concentrations could be explained by the assumption that all aerosol absorption is due to BC ( Schuster et al., 2005 [JoC] ), while a significant fraction may be due to absorption by organic aerosol and mineral dust (see Sections 2.4.4.2 ,and 2.4.4.6 ). Furthermore, Reddy et al. 2005a [JoC, SRC] ) show that comparison of the aerosol absorption τaer from models against those from AERONET must be performed very carefully, reducing the discrepancy between their model and AERONET derived aerosol absorption τaer from a factor of 4 to a factor of 1.2 by careful co-sampling of AERONET and model data. As mentioned above, uncertainty in the vertical position of absorbing aerosol relative to clouds can lead to large uncertainty in the TOA aerosol RF.
The partly absorbing nature of the aerosol is responsible for a heating of the lower-tropospheric column and also results in the surface forcing being considerably more negative than TOA RF, results that have been confirmed through several experimental and observational studies as discussed in earlier sections. Table 2.6 summarises the surface forcing obtained in the different models. Figure 2.12 depicts the regional distribution of several important parameters for assessing the regional impact of aerosol RF. The results are based on a mean model constructed from AeroCom simulation results B and PRE. Anthropogenic τaer ( Figure 2.12 a) is shown to have local maxima in industrialised regions and in areas dominated by biomass burning. The difference between simulated and observed τaer shows that regionally τaer can be up to 0.1( Figure 2.12 b). Figure 2.12 csuggests that there are regions off Southern Africa where the biomass burning aerosol above clouds leads to a local positive RF. Figure 2.12 dshows the local variability as the standard deviation from nine models of the overall RF. The largest uncertainties of ±3 Wm–2 are found in East Asia and in the African biomass burning regions. Figure 2.12 ereveals that an average of 0.9 Wm–2 heating can be expected in the atmospheric column as a consequence of absorption by anthropogenic aerosols. Regionally, this can reach annually averaged values exceeding 5 Wm–2.These regional effects and the negative surface forcing in the shortwave( Figure 2.12 f) is expected to exert an important effect on climate through alteration of the hydrological cycle.
An uncertainty estimate for the model-derived aerosol direct RF can be based upon two alternative error analyses:
1) An error propagation analysis using the errors given in the sections on sulphate, fossil fuel BC and organic carbon, biomass burning aerosol, nitrate and anthropogenic mineral dust. Assuming linear additivity of the errors, this results in an overall 90% confidence level uncertainty of 0.4 Wm–2.
2) The standard deviation of the aerosol direct RF results in Table 2.6 ,multiplied by 1.645, suggests a 90% confidence level uncertainty of 0.3 Wm–2,or 0.4 Wm–2 when mineral dust and nitrate aerosol are accounted for.
Therefore, the results summarised in Table 2.6 and Figure 2.13 ,together with the estimates of nitrate and mineral dust RF combined with the measurement-based estimates, provide an estimate for the combined aerosol direct RF of –0.50 ± 0.40 Wm–2.The progress in both global modelling and measurements of the direct RF of aerosol leads to a medium-low level of scientific understanding (see Section 2.9 , Table 2.11 ).
Table 2.6 .Quantities related to estimates of the aerosol direct RF. Recent estimates of anthropogenic aerosol load (LOAD), anthropogenic aerosol optical depth (τaer), its fraction of the present-day total aerosol optical depth (τaer ant), cloud cover in aerosol model, total aerosol direct radiative forcing (RF) for clear sky and all sky conditions, surface forcing and atmospheric all-sky forcing.
| No | Modela | LOAD | τaer (0.55 µm) | τaer ant (0.55 µm) | Cloud Cover | RF top clear sky | RF top all sky | Surface Forcing all sky | Atmospheric Forcing all sky | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| (mgm–2) | (%) | (%) | (Wm–2) | (Wm–2) | (Wm–2) | (Wm–2) | ||||
| Published since IPCC, 2001 | ||||||||||
| A | GISS | 5.0 | 79% | –0.39b +0.01c | –1.98b –2.42c | 1.59b 2.43c | (Liao and Seinfeld, 2005) | |||
| B | LOA | 6.0 | 0.049 | 34% | 70% | –0.53 | –0.09 | (Reddy and Boucher, 2004) | ||
| C | SPRINTARS | 4.8 | 0.044 | 50% | 63% | –0.77 | –0.06 | –1.92 | 1.86 | (Takemura et al., 2005) |
| D | UIO-GCM | 2.7 | 57% | –0.11 | (Kirkevag and Iversen, 2002) | |||||
| E | GATORG | 6.4d | 62% | –0.89 | –0.12 | –2.5 | 2.38 | (Jacobson, 2001a) | ||
| F | GISS | 6.7 | 0.049 | –0.23 | (Hansen et al., 2005) | |||||
| G | GISS | 5.6 | 0.040 | –0.63 | (Koch, 2001) | |||||
| AeroCom: identical emissions used for year 1750 and 2000 (Schulz et al., 2006) | ||||||||||
| H | UMI | 4.0 | 0.028 | 25% | 63% | –0.80 | –0.41 | –1.24 | 0.84 | (Liu and Penner, 2002) |
| I | UIO_CTM | 3.0 | 0.026 | 19% | 70% | –0.85 | –0.34 | –0.95 | 0.61 | (Myhre et al., 2003) |
| J | LOA | 5.3 | 0.046 | 28% | 70% | –0.80 | –0.35 | –1.49 | 1.14 | (Reddy and Boucher, 2004) |
| K | LSCE | 4.8 | 0.033 | 40% | 62% | –0.94 | –0.28 | –0.93 | 0.66 | (Schulz et al., 2006) |
| L | ECHAM5 | 4.3 | 0.032 | 30% | 62% | –0.64 | –0.27 | –0.98 | 0.71 | (Stier et al., 2005) |
| M | GISS | 2.8 | 0.014 | 11% | 57% | –0.29 | –0.11 | –0.81 | 0.79 | (Koch, 2001) |
| N | UIO_GCM | 2.8 | 0.017 | 11% | 57% | –0.01 | –0.84 | 0.84 | (Kirkevag and Iversen, 2002) | |
| O | SPRINTARS | 3.2 | 0.036 | 44% | 62% | –0.35 | +0.04 | –0.91 | 0.96 | (Takemura et al., 2005) |
| P | ULAQ | 3.7 | 0.030 | 23% | –0.79 | –0.24 | (Pitari et al., 2002) | |||
| Average A–G | 5.1 | 0.046 | 42% | 67% | –0.73 | –0.23 | –2.21 | 2.07 | ||
| Average H–P | 3.8 | 0.029 | 26% | 63% | –0.68 | –0.22 | –1.02 | 0.82 | ||
| Stddev A–G | 1.4 | 0.004 | 0.18 | 0.21 | ||||||
| Stddev H–P | 0.9 | 0.010 | 11% | 5% | 0.24 | 0.16 | 0.23 | 0.17 | ||
| Average A–P | 4.3 | 0.035 | 29% | 64% | –0.70 | –0.22 | –1.21 | 1.24 | ||
| Stddev A–P | 1.3 | 0.012 | 13% | 7% | 0.26 | 0.18 | 0.44 | 0.65 | ||
| Minimum A–P | 2.7 | 0.014 | 11% | 57% | –0.94 | –0.63 | –1.98 | 0.61 | ||
| Maximum A–P | 6.7 | 0.049 | 50% | 79% | –0.29 | 0.04 | –0.81 | 2.43 | ||
2.4.5 Aerosol Influence on Clouds (Cloud Albedo Effect)
As pointed out in Section 2.4.1 ,aerosol particles affect the formation and properties of clouds. Only a subset of the aerosol population acts as cloud condensation nuclei (CCN) and/or ice nuclei (IN). Increases in ambient concentrations of CCN and IN due to anthropogenic activities can modify the microphysical properties of clouds, thereby affecting the climate system ( Penner et al., 2001 [NPR, ARC] ; Ramanathan et al., 2001a [JoC, SRC] , Jacob et al., 2005 [NPR, MoS, ARC] ). Several mechanisms are involved, as presented schematically in Figure 2.10 .As noted in Ramaswamy et al. 2001 [NPR, MoS, SRC] ), enhanced aerosol concentrations can lead to an increase in the albedo of clouds under the assumption of fixed liquid water content ( Junge, 1975 [NPR, MoS] ; Twomey, 1977 [JoC] ); this mechanism is referred to in this report as the ‘cloud albedo effect’. The aerosol enhancements have also been hypothesised to lead to an increase in the lifetime of clouds ( Albrecht, 1989 [JoC] ); this mechanism is referred to in this report as the ‘cloud lifetime effect’ and discussed in Section 7.5 .
The interactions between aerosol particles (natural and anthropogenic in origin) and clouds are complex and can be nonlinear ( Ramaswamy et al., 2001 [NPR, MoS, SRC] ). The size and chemical composition of the initial nuclei (e.g., anthropogenic sulphates, nitrates, dust, organic carbon and BC) are important in the activation and early growth of the cloud droplets, particularly the water-soluble fraction and presence of compounds that affect surface tension ( McFiggans et al., 2006 [ARC] and references therein). Cloud optical properties are a function of wavelength. They depend on the characteristics of the droplet size distributions and ice crystal concentrations, and on the morphology of the various cloud types.
The interactions of increased concentrations of anthropogenic particles with shallow (stratocumulus and shallow cumulus) and deep convective clouds (with mixed phase) are discussed in this subsection. This section presents new observations and model estimates of the albedo effect. The associated RF in the context of liquid water clouds is assessed. In-depth discussion of the induced changes that are not considered as RFs (e.g., semi-direct and cloud cover and lifetime effects, thermodynamic response and changes in precipitation development) are presented in Section 7.5 .The impacts of contrails and aviation-induced cirrus are discussed in Section 2.6 and the indirect impacts of aerosol on snow albedo are discussed in Section 2.5.4 .
2.4.5.1 Link Between Aerosol Particles and Cloud Microphysics
The local impact of anthropogenic aerosols has been known for a long time. For example, smoke from sugarcane and forest fires was shown to reduce cloud droplet sizes in early case studies utilising in situ aircraft observations ( Warner and Twomey, 1967 [JoC] ; Eagan et al., 1974 [JoC] ). On a regional scale, studies have shown that heavy smoke from forest fires in the Amazon Basin have led to increased cloud droplet number concentrations and to reduced cloud droplet sizes ( Reid et al., 1999 [JoC, MoS] ; Andreae et al., 2004 [JoC] ; Mircea et al., 2005 [MoS] ). The evidence concerning aerosol modification of clouds provided by the ship track observations reported in the TAR has been further confirmed, to a large extent qualitatively, by results from a number of studies using in situ aircraft and satellite data, covering continental cases and regional studies. Twohy et al. 2005 [JoC] ) explored the relationship between aerosols and clouds in nine stratocumulus cases, indicating an inverse relationship between particle number and droplet size, but no correlation was found between albedo and particle concentration in the entire data set. Feingold et al. 2003 [JoC, MoS] Kim et al. 2003 [JoC, ARC] and Penner et al. 2004 [JoC, MoS, ARC] ) presented evidence of an increase in the reflectance in continental stratocumulus cases, utilising remote sensing techniques at specific field sites. The estimates in Feingold et al. 2003 [JoC, MoS] ) confirm that the relationship between aerosol and cloud droplet number concentrations is nonlinear, that is Nd≈ (Na)b,where Nd is the cloud drop number density and Na is the aerosol number concentration. The parameter b in this relationship can vary widely, with values ranging from 0.06 to 0.48 (low values of b correspond to low hygroscopicity). This range highlights the sensitivity to aerosol characteristics (primarily size distribution), updraft velocity and the usage of aerosol extinction as a proxy for CCN ( Feingold, 2003 [JoC, MoS] ). Disparity in the estimates of b (or equivalent) based on satellite studies ( Nakajima et al., 2001 [JoC, SRC] ; Breon et al., 2002 [JoC] ) suggests that a quantitative estimate of the albedo effect from remote sensors is problematic ( Rosenfeld and Feingold 2003 [JoC] ), particularly since measurements are not considered for similar liquid water paths.
Many recent studies highlight the importance of aerosol particle composition in the activation process and droplet spectral evolution (indicated in the early laboratory work of Gunn and Philips, 1957 ), but the picture that emerges is not complete. Airborne aerosol mass spectrometers provide firm evidence that ambient aerosols consist mostly of internal mixtures, for example, biomass burning components, organics and soot are mixed with other aerosol components ( McFiggans et al., 2006 [ARC] Mircea et al. 2005 [MoS] ) showed the importance of the organic aerosol fraction in the activation of biomass burning aerosol particles. The presence of internal mixtures (e.g., sea salt and organic compounds) can affect the uptake of water and the resulting optical properties compared to a pure sea salt particle ( Randles et al., 2004 [JoC, MoS, SRC] ). Furthermore, the varying contents of water-soluble and insoluble substances in internally mixed particles, the vast diversity of organics, and the resultant effects on cloud droplet sizes, makes the situation even more complex. Earlier observations of fog water ( Facchini et al., 1999 [JoC, ARC] , 2000 ) suggested that the presence of organic aerosols would reduce surface tension and lead to a significant increase in the cloud droplet number concentration ( Nenes et al., 2002 [JoC] ; Rissler et al., 2004 [MoS] ; Lohmann and Leck, 2005 [JoC, SRC] ; Ming et al., 2005a [JoC, MoS, SRC] ; McFiggans et al., 2006 [ARC] ). On the other hand, Feingold and Chuang 2002 [JoC] and Shantz et al. 2003 [JoC] ) indicated that organic coating on CCN delayed activation, leading to a reduction in drop number and a broadening of the cloud droplet spectrum, which had not been previously considered. Ervens et al. 2005 [JoC] ) addressed numerous composition effects in unison to show that the effect of composition on droplet number concentration is much less than suggested by studies that address individual composition effects, such as surface tension. The different relationships observed between cloud optical depth and liquid water path in clean and polluted stratocumulus clouds ( Penner et al., 2004 [JoC, MoS, ARC] ) have been explained by differences in sub-cloud aerosol particle distributions, while some contribution can be attributed to CCN composition (e.g., internally mixed insoluble dust; Asano et al., 2002 [JoC] ). Nevertheless, the review by McFiggans et al. 2006 [ARC] ) points to the remaining difficulties in quantitatively explaining the relationship between aerosol size and composition and the resulting droplet size distribution. Dusek et al. 2006 [JoC] ) concluded that the ability of a particle to act as a CCN is largely controlled by size rather than composition.
The complexity of the aerosol-cloud interactions and local atmospheric conditions where the clouds are developing are factors in the large variation evidenced for this phenomenon. Advances have been made in the understanding of the regional and/or global impact based on observational studies, particularly for low-level stratiform clouds that constitute a simpler cloud system to study than many of the other cloud types. Column aerosol number concentration and column cloud droplet concentration over the oceans from the AVHRR ( Nakajima et al., 2001 [JoC, SRC] ) indicated a positive correlation, and an increase in shortwave reflectance of low-level, warm clouds with increasing cloud optical thickness, while liquid water path (LWP) remained unmodified. While these results are only applicable over the oceans and are based on data for only four months, the positive correlation between an increase in cloud reflectance and an enhanced ambient aerosol concentration has been confirmed by other studies ( Brenguier et al., 2000a [JoC] ,b; Rosenfeld et al., 2002 [JoC] ). However, other studies highlight the sensitivity to LWP, linking high pollution entrained into clouds to a decrease in LWP and a reduction in the observed cloud reflectance ( Jiang et al., 2002 [JoC, MoS] ; Brenguier et al., 2003 [JoC] ; Twohy et al., 2005 [JoC] ). Still others ( Han et al., 2002 [JoC] , using AVHRR observations) have reported an absence of LWP changes in response to increases in the column-averaged droplet number concentration, this occurred for one-third of the cloud cases studied for which optical depths ranged between 1 and 15. Results of large-eddy simulations of stratocumulus ( Jiang et al., 2002 [JoC, MoS] ; Ackerman et al., 2004 [JoC, MoS] ; Lu and Seinfeld, 2005 [JoC, MoS] ) and cumulus clouds ( Jiang and Feingold, 2006 [JoC, MoS] ; Xue and Feingold, 2006 [JoC, MoS] ) seem to confirm the lack of increase in LWP due to increases in aerosols; they point to a dependence on precipitation rate and relative humidity above the clouds ( Ackerman et al., 2004 [JoC, MoS] ). The studies above highlight the difficulty of devising observational studies that can isolate the albedo effect from other effects (e.g., meteorological variability, cloud dynamics) that influence LWP and therefore cloud RF.
Results from the POLDER satellite instrument, which retrieves both submicron aerosol loading and cloud droplet size, suggest much larger cloud effective radii in remote oceanic regions than in the highly polluted continental source areas and downwind adjacent oceanic areas, namely from a maximum of 14 µm down to 6 µm ( Bréon et al., 2002 [JoC] ). This confirms earlier studies of hemispheric differences using AVHRR. Further, the POLDER- and AVHRR-derived correlations between aerosol and cloud parameters are consistent with an aerosol indirect effect ( Sekiguchi et al., 2003 [JoC] ). These results suggest that the impact of aerosols on cloud microphysics is global. Note that the satellite measurements of aerosol loading and cloud droplet size are not coincident, and an aerosol index is not determined in the presence of clouds. Further, there is a lack of simultaneous measurements of LWP, which makes assessment of the cloud albedo RF difficult.
The albedo effect is also estimated from studies that combined satellite retrievals with a CTM, for example, in the case of two pollution episodes over the mid-latitude Atlantic Ocean. Results indicated a brightening of clouds over a time scale of a few days in instances when LWP did not undergo any significant changes ( Hashvardhan et al., 2002 [JoC, MoS] ; Schwartz et al., 2002 [JoC, MoS, ARC] ; Krüger and Graβl, 2002 [NotFound] ). There have been fewer studies on aerosol-cloud relationships under more complex meteorological conditions (e.g., simultaneous presence of different cloud types).
The presence of insoluble particles within ice crystals constituting clouds formed at cold temperatures has a significant influence on the radiation transfer. The inclusions of scattering and absorbing particles within large ice crystals ( Macke et al., 1996 [JoC] ) suggest a significant effect. Hence, when soot particles are embedded, there is an increase in the asymmetry parameter and thus forward scattering. In contrast, inclusions of ammonium sulphate or air bubbles lead to a decrease in the asymmetry parameter of ice clouds. Given the recent observations of partially insoluble nuclei in ice crystals ( Cziczo et al., 2004 [JoC] ) and the presence of small crystal populations, there is a need to further develop the solution for radiative transfer through such systems.
2.4.5.2 Estimates of the Radiative Forcing from Models
General Circulation Models constitute an important and useful tool to estimate the global mean RF associated with the cloud albedo effect of anthropogenic aerosols. The model estimates of the changes in cloud reflectance are based on forward calculations, considering emissions of anthropogenic primary particles and secondary particle production from anthropogenic gases. Since the TAR, the cloud albedo effect has been estimated in a more systematic and rigorous way (allowing, for example, for the relaxation of the fixed LWC criterion), and more modelling results are now available. Most climate models use parametrizations to relate the cloud droplet number concentration to aerosol concentration; these vary in complexity from simple empirical fits to more physically based relationships. Some models are run under an increasing greenhouse gas concentration scenario and include estimates of present-day aerosol loadings (including primary and secondary aerosol production from anthropogenic sources). These global modelling studies( Table 2.7 )have a limitation arising from the underlying uncertainties in aerosol emissions (e.g., emission rates of primary particles and of secondary particle precursors). Another limitation is the inability to perform a meaningful comparison between the various model results owing to differing formulations of relationships between aerosol particle concentrations and cloud droplet or ice crystal populations; this, in turn, yields differences in the impact of microphysical changes on the optical properties of clouds. Further, even when the relationships used in different models are similar, there are noticeable differences in the spatial distributions of the simulated low-level clouds. Individual models’ physics have undergone considerable evolution, and it is difficult to clearly identify all the changes in the models as they have evolved. While GCMs have other well-known limitations, such as coarse spatial resolution, inaccurate representation of convection and hence updraft velocities leading to aerosol activation and cloud formation processes, and microphysical parametrizations, they nevertheless remain an essential tool for quantifying the global cloud albedo effect. In Table 2.7 ,differences in the treatment of the aerosol mixtures (internal or external, with the latter being the more frequently employed method) are noted. Case studies of droplet activation indicate a clear sensitivity to the aerosol composition ( McFiggans et al., 2006 [ARC] ); additionally, radiative transfer is sensitive to the aerosol composition and the insoluble fraction present in the cloud droplets.
Table 2.7 .Published model studies of the RF due to cloud albedo effect, in the context of liquid water clouds, with a listing of the relevant modelling details.
| Model | Model typea | Aerosol speciesb | Aerosol mixturesc | Cloud types included | Microphysics | Radiative Forcing(Wm–2)d |
|---|---|---|---|---|---|---|
| Lohmann et al. (2000) | AGCM + sulphur cycle (ECHAM4) | S, OC, BC, SS, D | I | warm and mixed phase |
Droplet number concentration and LWC, Beheng (1994); Sundqvist et al. (1989). Also, mass and number from field observations
|
–1.1 (total) –0.45 (albedo) |
| E |
–1.5 (total) | |||||
| Jones et al. (2001) | AGCM + sulphur cycle, fixed SST (Hadley) | S, SS, D (a crude attempt for D over land, no radiation) | E | stratiform and shallow cumulus |
Droplet number concentration and LWC, Wilson and Ballard (1999); Smith (1990); Tripoli and Cotton (1980); Bower et al. (1994). Warm and mixed phase, radiativetreatment of anvil cirrus, non-spherical ice particles |
–1.89 (total) –1.34 (albedo) |
| Williams et al. (2001b) | GCM with slab ocean + sulphur cycle (Hadley) | S, SS | E | stratiform and shallow cumulus |
Jones et al. (2001)
|
–1.69 (total) –1.37 (albedo) |
| AGCM, fixed SST |
–1.62 (total) –1.43(albedo) | |||||
| Rotstayn and Penner (2001) | AGCM (CSIRO), fixed SST and sulphur loading | S | n.a. | warm and mixed phase |
Rotstayn (1997); Rotstayn et al. (2000) |
–1.39 (albedo) |
| Rotstayn and Liu (2003) value decreased) | Interactive sulphur cycle |
Inclusion of dispersion |
12 to 35% decrease –1.12 (albedo, mid value decreased) | |||
| Ghan et al. (2001) | AGCM (PNNL) + chemistry (MIRAGE), fixed SST | S, OC, BC, SS, N, D | E(for different modes); I (within modes) | warm and mixed phase |
Droplet number concentration and LWC, crystal concentration and ice water content. Different processes affecting the various modes |
–1.7 (total) –0.85 (albedo) |
| Chuang et al. (2002) | CCM1 (NCAR) + chemistry (GRANTOUR), fixed SST | S, OC, BC, SS, D | E(for emitted particles); I: when growingby condensation | warm and mixed phase |
Modified from Chuang and Penner (1995), no collision/coalescence |
–1.85 (albedo) |
| Menon et al. (2002a) | GCM (GISS) + sulphur cycle, fixed SST | S,OC, SS | E | warm |
Droplet number concentration and LWC, Del Genio et al. (1996), Sundqvist et al. (1989). Warm and mixed phase, improved vertical distribution of clouds (but only ninelayers).Global aerosol burdens poorly constrained |
–2.41 (total) –1.55 (albedo) |
| Kristjansson (2002) | CCM3 (NCAR) fixed SST | S, OC, BC, SS, D | E(for nucleation mode and fossil fuel BC); I (foraccumulationmode) | warm and mixed phase |
Rasch and Kristjánsson (1998). Stratiform and detraining convective clouds |
–1.82 (total) –1.35 (albedo) |
| Suzuki et al. (2004) | AGCM (Japan), fixed SST | S, OC, BC, SS | E | stratiform |
Berry(1967), Sundqvist(1978) |
0.54 (albedo) |
| Quaas et al. (2004) | AGCM (LMDZ) + interactive sulphur cycle, fixed SST | S | n.a. | warm and mixed phase |
Aerosol mass and cloud droplet number concentration, Boucher and Lohmann (1995); Boucher et al. (1995) |
–1.3 (albedo) |
| Hansen et al. (2005) shallow (below | GCM (GISS) + 3 different ocean parametrizations | S, OC, BC, SS, N, D (D not included in clouds) | E | warm and 720hPa) |
Schmidt et al. (2005), 20 vertical layers. Droplet number concentration (Menon and Del Genio, 2007) |
–0.77 (albedo) |
| Kristjansson et al. (2005) | CCM3 (NCAR) + sulphur and carbon cycles slab ocean | S, OC, BC, SS, D | E(for nucleation mode and fossil fuel BC); I (foraccumulationmode) | warm and mixed phase |
Kristjansson (2002). Stratiform and detraining convective clouds |
–1.15 (total,at the surface) |
| Quaas and Boucher (2005) | AGCM (LMDZ) + interactive sulphur cycle, fixed SST | S, OC, BC, SS, D | E | warm and mixed phase |
Aerosol mass and cloud droplet number concentration, Boucher and Lohmann (1995); Boucher et al. (1995)control run |
–0.9 (albedo) |
|
fit to POLDER data fit to MODIS data |
–0.5 (albedo)e –0.3 (albedo)e | |||||
| Quaas et al. (2005) | AGCM (LMDZ and ECHAM4) | S, OC, BC, SS, D | E | warm and mixed phase |
Aerosol mass and cloud droplet number concentration, Boucher and Lohmann, (1995), control runs (ctl) |
–0.84 (total LMDZ-ctl) –1.54 (total(ECHAM4-ctl) |
|
Aerosol mass and cloud droplet number concentration fitted to MODIS data |
–0.53 (total LMDZ)e –0.29 (total(ECHAM4)e | |||||
| Dufresne et al. (2005) | AGCM (LMDZ) + interactive sulphur cycle, fixed SST | S | n.a. | warm |
Aerosol mass and cloud droplet number concentration, Boucher and Lohmann, (1995), fitted to POLDER data |
–0.22 (albedo)e |
| Takemura et al. (2005) | AGCM (SPRINTARS) + slab ocean | S, OC, BC, SS, D | E(50% BC from fossil fuel); I (for OC and BC) | warm |
Activation based on Kohler theory and updraft velocity |
–0.94 (total) –0.52 (albedo) |
| Chen and Penner (2005) | AGCM (UM) + fixed SST | S, SS, D, OC, BC | I | warm and mixed phase |
Aerosol mass and cloud droplet number concentration(lognormal)Control (Abdul-Razzak and Ghan, 2002) |
–1.30 (albedo,UM_ctrl)f |
|
Relationship between droplet concentration and dispersion coefficient: High |
–0.75 (albedo,UM_1)f | |||||
|
Relationship between droplet concentration and dispersion coefficient: Medium Updraft velocity |
–0.86 (albedo,UM_2)f –1.07 (albedo,UM_3)f | |||||
|
Relationship between droplet concentration and dispersion coefficient: Low Chuang et al. (1997) |
–1.10 (albedo, UM_4)f –1.29 (albedo,UM_5)f | |||||
|
Nenes and Seinfeld (2003) |
–1.79 (albedo,UM_6)f | |||||
| Ming et al. (2005b) | AGCM (GFDL), fixed SST and sulphur loading | S | n.a. | warm |
Rotstayn et al. (2000), Khainroutdinov and Kogan (2000). Aerosols off-line |
–2.3 (total) –1.4 (albedo) |
| Penner et al. (2006) results from experiment 1 | LMDZ, Oslo and CCSR | S, SS, D, OC, BC | E | warm and mixed phase |
Aerosol mass and cloud droplet number concentration; Boucher and Lohmann, (1995); Chen and Penner (2005); Sundqvist (1978) |
–0.65 (albedo Oslo) –0.68 (albedo LMDZ) –0.74 (albedo CCSR) |
All models estimate a negative global mean RF associated with the cloud albedo effect, with the range of model results varying widely, from –0.22 to –1.85 Wm–2.There are considerable differences in the treatment of aerosol, cloud processes and aerosol-cloud interaction processes in these models. Several models include an interactive sulphur cycle and anthropogenic aerosol particles composed of sulphate, as well as naturally produced sea salt, dust and continuously outgassing volcanic sulphate aerosols. Lohmann et al. 2000 [JoC, SRC] and Chuang et al. 2002 [JoC, MoS] ) included internally mixed sulphate, black and organic carbon, sea salt and dust aerosols, resulting in the most negative estimate of the cloud albedo indirect effect. Takemura et al. 2005 [JoC, MoS, SRC] ) used a global aerosol transport-radiation model coupled to a GCM to estimate the direct and indirect effects of aerosols and their associated RF. The model includes a microphysical parametrization to diagnose the cloud droplet number concentration using Köhler theory, which depends on the aerosol particle number concentration, updraft velocity, size distributions and chemical properties of each aerosol species. The results indicate a global decrease in cloud droplet effective radius caused by anthropogenic aerosols, with the global mean RF calculated to be –0.52 Wm–2;the land and oceanic contributions are –1.14 and –0.28 Wm–2,respectively. Other modelling results also indicate that the mean RF due to the cloud albedo effect is on average somewhat larger over land than over oceans; over oceans there is a more consistent response from the different models, resulting in a smaller inter-model variability ( Lohmann and Feichter, 2005 [SRC] ).
Chen and Penner 2005 [ARC] ), by systematically varying parameters, obtained a less negative RF when the in-cloud updraft velocity was made to depend on the turbulent kinetic energy. Incorporating other cloud nucleation schemes, for example, changing from Abdul-Razzak and Ghan 2002 [JoC] ) to the Chuang et al. 1997 [JoC] ) parametrization resulted in no RF change, while changing to the Nenes and Seinfeld 2003 [NPR, JoC, MoS] ) parametrization made the RF more negative. Rotstayn and Liu 2003 [JoC, ARC] ) found a 12 to 35% decrease in the RF when the size dispersion effect was included in the case of sulphate particles. Chen and Penner 2005 [ARC] ) further explored the range of parameters used in Rotstayn and Liu 2003 [JoC, ARC] ) and found the RF to be generally less negative than in the standard integration.
Amodel intercomparison study ( Penner et al., 2006 [MoS, ARC] ) examined the differences in cloud albedo effect between models through a series of controlled experiments that allowed examination of the uncertainties. This study presented results from three models, which were run with prescribed aerosol mass-number concentration (from Boucher and Lohmann, 1995 [JoC, MoS, SRC] ), aerosol field (from Chen and Penner, 2005 [ARC] ) and precipitation efficiency (from Sundqvist, 1978 [JoC, MoS] ). The cloud albedo RFs in the three models do not vary widely: –0.65, –0.68 and –0.74 Wm–2,respectively. Nevertheless, changes in the autoconversion scheme led to a differing response of the LWP between the models, and this is identified as an uncertainty.
Acloser inspection of the treatment of aerosol species in the models leads to a broad separation of the results into two groups: models with only a few aerosol species and those that include a more complex mixture of aerosols of different composition. Thus, in Figure 2.14 ,RF results are grouped according to the type of aerosol species included in the simulations. In the top panel of Figure 2.14 ,which shows estimates from models that mainly include anthropogenic sulphate, there is an indication that the results are converging, even though the range of models comes from studies published between 2001 and 2006 . These studies show much less scatter than in the TAR, with a mean and standard deviation of –1.37 ± 0.14 Wm–2.In contrast, in the bottom panel of Figure 2.14 ,which shows the studies that include more species, a much larger variability is found. These latter models (see Table 2.7 )include ‘state of the art’ parametrizations of droplet activation for a variety of aerosols, and include both internal and external mixtures.
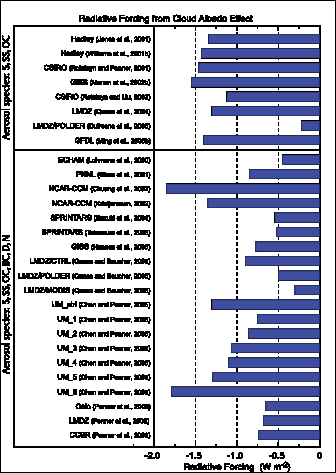
Figure 2.14. Radiative forcing due to the cloud albedo effect, in the context of liquid water clouds, from the global climate models that appear in Table 2.7 .The labels next to the bars correspond to the published study; the notes of Table 2.7 explain the species abbreviations listed on the left hand side. Top panel: results for models that consider a limited number of species, primarily anthropogenic sulphate (S). Bottom panel: results from studies that include a variety of aerosol compositions and mixtures; the estimates here cover a larger range than those in the top panel. Chen and Penner 2005 [ARC] ) presented a sensitivity study obtained by changing parametrizations in their model, so the results can be considered independent and are thus listed separately. Penner et al. 2006 [MoS, ARC] ) is an intercomparison study, so the results of the individual models are listed separately.
Some studies have commented on inconsistencies between some of the earlier estimates of the cloud albedo RF from forward and inverse calculations ( Anderson et al., 2003 [JoC, MoS] ). Notwithstanding the fact that these two streams of calculations rely on very different formulations, the results here appear to be within range of the estimates from inverse calculations.
2.4.5.3 Estimates of the Radiative Forcing from Observations and Constrained Models
It is difficult to obtain a best estimate of the cloud albedo RF from pre-industrial times to the present day based solely on observations. The satellite record is not long enough, and other long-term records do not provide the pre-industrial aerosol and cloud microphysical properties needed for such an assessment. Some studies have attempted to estimate the RF by incorporating empirical relationships derived from satellite observations. This approach is valid as long as the observations are robust, but problems still remain, particularly with the use of the aerosol optical depth as proxy for CCN ( Feingold et al., 2003 [JoC, MoS] ), droplet size and cloud optical depth from broken clouds ( Marshak et al., 2006 [JoC] ), and relative humidity effects ( Kapustin et al., 2006 [JoC] ) to discriminate between hydrated aerosols and cloud. Radiative forcing estimates constrained by satellite observations need to be considered with these caveats in mind.
By assuming a bimodal lognormal size distribution, Nakajima et al. 2001 [JoC, SRC] ) determined the Ångstrom exponent from AVHRR data over the oceans (for a period of four months), together with cloud properties, optical thickness and effective radii. The nonlinear relationship between aerosol number concentration and cloud droplet concentration(Nd≈ (Na)b)obtained is consistent with Twomey’s hypothesis; however, the parameter b is smaller than previous estimates (0.5 compared with 0.7 to 0.8; Kaufman et al., 1991 [JoC] ), but larger than the 0.26 value obtained by Martin et al. 1994 [JoC] ). Using this relationship, Nakajima et al. 2001 [JoC, SRC] ) provided an estimate of the cloud albedo RF in the range between –0.7 and –1.7 Wm–2,with a global average of –1.3 Wm–2 .Lohmann and Lesins 2002 [JoC, SRC] ) used POLDER data to estimate aerosol index and cloud droplet radius; they then scaled the results of the simulations with the European Centre Hamburg (ECHAM4) model. The results show that changes in Nalead to larger changes in Nd in the model than in observations, particularly over land, leading to an overestimate of the cloud albedo effect. The scaled values using the constraint from POLDER yield a global cloud albedo RF of –0.85 Wm–2,an almost 40% reduction from their previous estimate. Sekiguchi et al. 2003 [JoC] ) presented results from the analysis of AVHRR data over the oceans, and of POLDER data over land and ocean. Assuming that the aerosol column number concentration increased by 30% from the pre-industrial era, they estimated the effect due to the aerosol influence on clouds as the difference between the forcing under present and pre-industrial conditions. They estimated a global effect due to the total aerosol influence on clouds (sum of cloud albedo and lifetime effects) to be between –0.6 and –1.2 Wm–2,somewhat lower than the Nakajima et al. 2001 [JoC, SRC] ) ocean estimate. When the assumption is made that the liquid water content is constant, the cloud albedo RF estimated from AVHRR data is –0.64 ± 0.16 Wm–2 and the estimate using POLDER data is –0.37 ± 0.09 Wm–2.The results from these two studies are very sensitive to the magnitude of the increase in the aerosol concentration from pre-industrial to current conditions, and the spatial distributions.
Quaas and Boucher 2005 [JoC, MoS, SRC] ) used the POLDER and MODIS data to evaluate the relationship between cloud properties and aerosol concentrations on a global scale in order to incorporate it in a GCM. They derived relationships corresponding to marine stratiform clouds and convective clouds over land that show a decreasing effective radius as the aerosol optical depth increases. These retrievals involve a variety of assumptions that introduce uncertainties in the relationships, in particular the fact that the retrievals for aerosol and cloud properties are not coincident and the assumption that the aerosol optical depth can be linked to the sub-cloud aerosol concentration. When these empirical parametrizations are included in a climate model, the simulated RF due to the cloud albedo effect is reduced by 50% from their baseline simulation. Quaas et al. 2005 [Ambiguous] ) also utilised satellite data to establish a relationship between cloud droplet number concentration and fine-mode aerosol optical depth, minimising the dependence on cloud liquid water content but including an adiabatic assumption that may not be realistic in many cases. This relationship is implemented in the ECHAM4 and Laboratoire de Météorologie Dynamique Zoom (LMDZ) climate models and the results indicate that the original parametrizations used in both models overestimated the magnitude of the cloud albedo effect. Even though both models show a consistent weakening of the RF, it should be noted that the original estimates of their respective RFs are very different (by almost a factor of two); the amount of the reduction was 37% in LMDZ and 81% in ECHAM4. Note that the two models have highly different spatial distributions of low clouds, simulated aerosol concentrations and anthropogenic fractions.
When only sulphate aerosols were considered, Dufresne et al. 2005 [JoC] ) obtained a weaker cloud albedo RF. Their model used a relationship between aerosol mass concentration and cloud droplet number concentration, modified from that originally proposed by Boucher and Lohmann 1995 [JoC, MoS, SRC] ) and adjusted to POLDER data. Their simulations give a factor of two weaker RF compared to the previous parametrization, but it is noted that the results are highly sensitive to the distribution of clouds over land.
2.4.5.4 Uncertainties in Satellite Estimates
The improvements in the retrievals and satellite instrumentation have provided valuable data to begin observation-motivated assessments of the effect of aerosols on cloud properties, even though satellite measurements cannot unambiguously distinguish natural from anthropogenic aerosols. Nevertheless, an obvious advantage of the satellite data is their global coverage, and such extensive coverage can be analysed to determine the relationships between aerosol and cloud properties at a number of locations around the globe. Using these data some studies ( Sekiguchi et al., 2003 [JoC] ; Quaas et al., 2004 [JoC, MoS, SRC] ) indicate that the magnitude of the RF is resolution dependent, since the representation of convection and clouds in the GCMs and the simulation of updraft velocity that affects activation themselves are resolution dependent. The rather low spatial and temporal resolution of some of the satellite data sets can introduce biases by failing to distinguish aerosol species with different properties. This, together with the absence of coincident LWP measurements in several instances, handicaps the inferences from such studies, and hinders an accurate analysis and estimate of the RF. Furthermore, the ability to separate meteorological from chemical influences in satellite observations depends on the understanding of how clouds respond to meteorological conditions.
Retrievals involve a variety of assumptions that introduce uncertainties in the relationships. As mentioned above, the retrievals for aerosol and cloud properties are not coincident and the assumption is made that the aerosol optical depth can be linked to the aerosol concentration below the cloud. The POLDER instrument may underestimate the mean cloud-top droplet radius due to uncertainties in the sampling of clouds ( Rosenfeld and Feingold, 2003 [JoC] ). The retrieval of the aerosol index over land may be less reliable and lead to an underestimate of the cloud albedo effect over land. There is an indication of a systematic bias between MODIS-derived cloud droplet radius and that derived from POLDER ( (Breon and Doutriaux-Boucher, 2005 ) ), as well as differences in the aerosol optical depth retrieved from those instruments ( Myhre et al., 2004a [JoC, SRC] ) that need to be resolved.
2.4.5.5 Uncertainties Due to Model Biases
One of the large sources of uncertainties is the poor knowledge of the amount and distribution of anthropogenic aerosols used in the model simulations, particularly for pre-industrial conditions. Some studies show a large sensitivity in the RF to the ratio of pre-industrial to present-day aerosol number concentrations.
All climate models discussed above include sulphate particles; some models produce them from gaseous precursors over oceans, where ambient concentrations are low, while some models only condense mass onto pre-existing particles over the continents. Some other climate models also include sea salt and dust particles produced naturally, typically relating particle production to wind speed. Some models include anthropogenic nitrate, BC and organic compounds, which in turn affect activation. Models also have weaknesses in representing convection processes and aerosol distributions, and simulating updraft velocities and convection-cloud interactions. Even without considering the existing biases in the model-generated clouds, differences in the aerosol chemical composition and the subsequent treatment of activation lead to uncertainties that are difficult to quantify and assess. The presence of organic carbon, owing to its distinct hygroscopic and absorption properties, can be particularly important for the cloud albedo effect in the tropics ( Ming et al., 2007 [JoC, MoS, 2007] ).
Modelling the cloud albedo effect from first principles has proven difficult because the representation of aerosol-cloud and convection-cloud interactions in climate models are still crude ( Lohmann and Feichter, 2005 [SRC] ). Clouds often do not cover a complete grid box and are inhomogeneous in terms of droplet concentration, effective radii and LWP, which introduces added complications in the microphysical and radiative transfer calculations. Model intercomparisons (e.g., Lohmann et al., 2001 [JoC, MoS, SRC] ; Menon et al., 2003 [JoC, MoS, ARC] ) suggest that the predicted cloud distributions vary significantly between models, particularly their horizontal and vertical extents; also, the vertical resolution and parametrization of convective and stratiform clouds are quite different between models ( Chen and Penner, 2005 [ARC] ). Even high-resolution models have difficulty in accurately estimating the amount of cloud liquid and ice water content in a grid box.
It has proven difficult to compare directly the results from the different models, as uncertainties are not well identified and quantified. All models could be suffering from similar biases, and modelling studies do not often quote the statistical significance of the RF estimates that are presented. Ming et al. 2005b [JoC, MoS] ) demonstrated that it is only in the mid-latitude NH that their model yields a RF result at the 95% confidence level when compared to the unforced model variability. There are also large differences in the way that the different models treat the appearance and evolution of aerosol particles and the subsequent cloud droplet formation. Differences in the horizontal and vertical resolution introduce uncertainties in their ability to accurately represent the shallow warm cloud layers over the oceans that are most susceptible to the changes due to anthropogenic aerosol particles. A more fundamental problem is that GCMs do not resolve the small scales (order of hundreds of metres) at which aerosol-cloud interactions occur. Chemical composition and size distribution spectrum are also likely insufficiently understood at a microphysical level, although some modelling studies suggest that the albedo effect is more sensitive to the size than to aerosol composition ( Feingold, 2003 [JoC, MoS] ; Ervens et al., 2005 [JoC] ; Dusek et al., 2006 [JoC] ). Observations indicate that aerosol particles in nature tend to be composed of several compounds and can be internally or externally mixed. The actual conditions are difficult to simulate and possibly lead to differences among climate models. The calculation of the cloud albedo effect is sensitive to the details of particle chemical composition (activation) and state of the mixture (external or internal). The relationship between ambient aerosol particle concentrations and resulting cloud droplet size distribution is important during the activation process; this is a critical parametrization element in the climate models. It is treated in different ways in different models, ranging from simple empirical functions ( Menon et al., 2002a [JoC, MoS, ARC] ) to more complex physical parametrizations that also tend to be more computationally costly ( Abdul-Razzak and Ghan, 2002 [JoC] ; Nenes and Seinfeld, 2003 [NPR, JoC, MoS] ; Ming et al., 2006 [JoC, MoS, SRC] ). Finally, comparisons with observations have not yet risen to the same degree of verification as, for example, those for the direct RF estimates; this is not merely due to model limitations, since the observational basis also has not yet reached a sound footing.
Further uncertainties may be due to changes in the droplet spectral shape, typically considered invariant in climate models under clean and polluted conditions, but which can be substantially different in typical atmospheric conditions (e.g., Feingold et al., 1997 [JoC, MoS] ; Ackerman et al., 2000b [JoC] ; Erlick et al., 2001 [JoC, SRC] ; Liu and Daum, 2002 [JoC, MoS] Liu and Daum 2002 [JoC, MoS] ) estimated that a 15% increase in the width of the size distribution can lead to a reduction of between 10 and 80% in the estimated RF of the cloud albedo indirect effect. Peng and Lohmann 2003 [JoC, SRC] Rotstayn and Liu 2003 [JoC, ARC] and Chen and Penner 2005 [ARC] ) studied the sensitivity of their estimates to this dispersion effect. These studies confirm that their estimates of the cloud albedo RF, without taking the droplet spectra change into account, are overestimated by about 15 to 35%.
The effects of aerosol particles on heterogeneous ice formation are currently insufficiently understood and present another level of challenge for both observations and modelling. Ice crystal concentrations cannot be easily measured with present in situ instrumentation because of the difficulty of detecting small particles ( (Hirst et al., 2001 ) ) and frequent shattering of ice particles on impact with the probes ( (Korolev and Isaac, 2005 ) ). Current GCMs do not have sufficiently rigorous microphysics or sub-grid scale processes to accurately predict cirrus clouds or super-cooled clouds explicitly. Ice particles in clouds are often represented by simple shapes (e.g., spheres), even though it is well known that few ice crystals are like that in reality. The radiative properties of ice particles in GCMs often do not effectively simulate the irregular shapes that are normally found, nor do they simulate the inclusions of crustal material or soot in the crystals.
2.4.5.6 Assessment of the Cloud Albedo Radiative Forcing
As in the TAR, only the aerosol interaction in the context of liquid water clouds is assessed, with knowledge of the interaction with ice clouds deemed insufficient. Since the TAR, the cloud albedo effect has been estimated in a more systematic way, and more modelling results are now available. Models now are more advanced in capturing the complexity of the aerosol-cloud interactions through forward computations. Even though major uncertainties remain, clear progress has been made, leading to a convergence of the estimates from the different modelling efforts. Based on the results from all the modelling studies shown in Figure 2.14 ,compared to the TAR it is now possible to present a best estimate for the cloud albedo RF of –0.7 Wm–2 as the median, with a 5 to 95% range of –0.3 to –1.8 Wm–2.The increase in the knowledge of the aerosol-cloud interactions and the reduction in the spread of the cloud albedo RF since the TAR result in an elevation of the level of scientific understanding to low( Section 2.9 , Table 2.11 ).
2.5 Anthropogenic Changes in Surface Albedo and the Surface Energy Budget
2.5.1 Introduction
Anthropogenic changes to the physical properties of the land surface can perturb the climate, both by exerting an RF and by modifying other processes such as the fluxes of latent and sensible heat and the transfer of momentum from the atmosphere. In addition to contributing to changes in greenhouse gas concentrations and aerosol loading, anthropogenic changes in the large-scale character of the vegetation covering the landscape (‘land cover’) can affect physical properties such as surface albedo. The albedo of agricultural land can be very different from that of a natural landscape, especially if the latter is forest. The albedo of forested land is generally lower than that of open land because the greater leaf area of a forest canopy and multiple reflections within the canopy result in a higher fraction of incident radiation being absorbed. Changes in surface albedo induce an RF by perturbing the shortwave radiation budget ( Ramaswamy et al., 2001 [NPR, MoS, SRC] ). The effect is particularly accentuated when snow is present, because open land can become entirely snow-covered and hence highly reflective, while trees can remain exposed above the snow ( Betts, 2000 [JoC, SRC] ). Even a snow-covered canopy exhibits a relatively low albedo as a result of multiple reflections within the canopy ( Harding and Pomeroy, 1996 [JoC] ). Surface albedo change may therefore provide the dominant influence of mid- and high-latitude land cover change on climate ( Betts, 2001 [MoS, SRC] ; Bounoua et al., 2002 [JoC] ). The TAR cited two estimates of RF due to the change in albedo resulting from anthropogenic land cover change relative to potential natural vegetation (PNV), –0.4 Wm–2 and –0.2 Wm–2,and assumed that the RF relative to 1750 was half of that relative to PNV, so gave a central estimate of the RF due to surface albedo change of –0.2 Wm–2 ± 0.2 Wm–2.
Surface albedo can also be modified by the settling of anthropogenic aerosols on the ground, especially in the case of BC on snow ( Hansen and Nazarenko, 2004 [JoC, MoS, ARC] ). This mechanism may be considered an RF mechanism because diagnostic calculations may be performed under the strict definition of RF (see Sections 2.2 and 2.8 ). This mechanism was not discussed in the TAR.
Land cover change can also affect other physical properties such as surface emissivity, the fluxes of moisture through evaporation and transpiration, the ratio of latent to sensible heat fluxes (the Bowen ratio) and the aerodynamic roughness, which exerts frictional drag on the atmosphere and also affects turbulent transfer of heat and moisture. All these processes can affect the air temperature near the ground, and also modify humidity, precipitation and wind speed. Direct human perturbations to the water cycle, such as irrigation, can affect surface moisture fluxes and hence the surface energy balance. Changes in vegetation cover can affect the production of dust, which then exerts an RF. Changes in certain gases, particularly CO2 and ozone, can also exert an additional influence on climate by affecting the Bowen ratio, through plant responses that affect transpiration. These processes are discussed in detail in Section 7.2 .While such processes will act as anthropogenic perturbations to the climate system ( Pielke et al., 2002 [JoC] ) and will fall at least partly within the ‘forcing’ component of the forcing-feedback-response conceptual model, it is difficult to unequivocally quantify the pure forcing component as distinct from feedbacks and responses. The term ‘non-radiative forcing’ has been proposed ( Jacob et al., 2005 [NPR, MoS, ARC] ) and this report adopts the similar term ‘non-initial radiative effect’, but no quantitative metric separating forcing from feedback and response has yet been implemented for climatic perturbation processes that do not act directly on the radiation budget (see Section 2.2 ).
Energy consumption by human activities, such as heating buildings, powering electrical appliances and fuel combustion by vehicles, can directly release heat into the environment. This was not discussed in the TAR. Anthropogenic heat release is not an RF, in that it does not directly perturb the radiation budget; the mechanisms are not well identified and so it is here referred to as a non-initial radiative effect. It can, however, be quantified as a direct input of energy to the system in terms of Wm–2.
2.5.2 Changes in Land Cover Since 1750
In 1750, 7.9 to 9.2 million km2 (6 to 7% of the global land surface) were under cultivation or pasture( Figure 2.15 ), mainly in Europe, the Indo-Gangetic Plain and China ( Ramankutty and Foley, 1999 [JoC, MoS, SRC] ; Klein Goldewijk, 2001 [JoC, MoS] ). Over the next hundred years, croplands and pasture expanded and intensified in these areas, and new agricultural areas emerged in North America. The period 1850 to 1950 saw a more rapid rate of increase in cropland and pasture areas. In the last 50 years, several regions of the world have seen cropland areas stabilise, and even decrease. In the USA, as cultivation shifted from the east to the Midwest, croplands were abandoned along the eastern seaboard around the turn of the century and the eastern forests have regenerated over the last century. Similarly, cropland areas have decreased in China and Europe. Overall, global cropland and pasture expansion was slower after 1950 than before. However, deforestation is occurring more rapidly in the tropics. Latin America, Africa and South and Southeast Asia experienced slow cropland expansion until the 20th century, but have had exponential increases in the last 50 years. By 1990, croplands and pasture covered 45.7 to 51.3 million km2 (35% to 39% of global land), and forest cover had decreased by roughly 11 million km2 ( Ramankutty and Foley, 1999 [JoC, MoS, SRC] ; Klein Goldewijk, 2001 [JoC, MoS] ; Table 2.8 ).
Overall, until the mid-20th century most deforestation occurred in the temperate regions( Figure 2.15 ). In more recent decades, however, land abandonment in Western Europe and North America has been leading to reforestation while deforestation is now progressing rapidly in the tropics. In the 1990 s compared to the 1980 s, net removal of tropical forest cover had slowed in the Americas but increased in Africa and Asia.
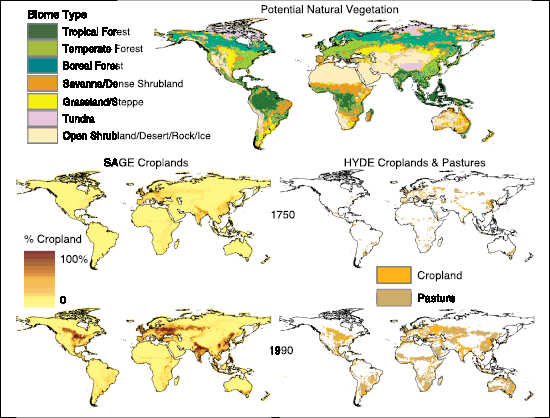
Figure 2.15. Anthropogenic modifications of land cover up to 1990 . Top panel: Reconstructions of potential natural vegetation ( Haxeltine and Prentice, 1996 [JoC, MoS] ). Lower panels: reconstructions of croplands and pasture for 1750 and 1990 . Bottom left: fractional cover of croplands from Centre for Sustainability and the Global Environment (SAGE; Ramankutty and Foley, 1999 [JoC, MoS, SRC] ) at 0.5° resolution. Bottom right: reconstructions from the HistorY Database of the Environment (HYDE; Klein Goldewijk, 2001 [JoC, MoS] ), with one land cover classification per 0.5° grid box.
2.5.3 Radiative Forcing by Anthropogenic Surface Albedo Change: Land Use
Since the TAR, a number of estimates of the RF from land use changes over the industrial era have been made( Table 2.8 ). Unlike the main TAR estimate, most of the more recent studies are ‘pure’ RF calculations with the only change being land cover; feedbacks such as changes in snow cover are excluded. Brovkin et al. 2006 [JoC, MoS, ARC] ) estimated the global mean RF relative to 1700 to be –0.15 Wm–2,considering only cropland changes ( Ramankutty and Foley, 1999 [JoC, MoS, SRC] ) and not pastures. Hansen et al. 2005 [JoC, MoS, ARC] ) also considered only cropland changes ( Ramankutty and Foley, 1999 [JoC, MoS, SRC] ) and simulated the RF relative to 1750 to be –0.15 Wm–2.Using historical reconstructions of both croplands ( Ramankutty and Foley, 1999 [JoC, MoS, SRC] ) and pasturelands ( Klein Goldewijk, 2001 [JoC, MoS] Betts et al. 2007 [MoS, SRC, 2007] ) simulated an RF of –0.18 Wm–2 since 1750 . This study also estimated the RF relative to PNV to be –0.24 Wm–2.Other studies since the TAR have also estimated the RF at the present day relative to PNV( Table 2.8 Govindasamy et al. 2001a [JoC] ) estimated this RF as –0.08 Wm–2 .Myhre et al. 2005a [JoC, MoS, SRC] ) used land cover and albedo data from MODIS ( (Friedl et al., 2002; ) ( Schaaf et al., 2002 ) ) and estimated this RF as –0.09 Wm–2.The results of Betts et al. 2007 [MoS, SRC, 2007] and Brovkin et al. 2006 [JoC, MoS, ARC] ) suggest that the RF relative to 1750 is approximately 75% of that relative to PNV. Therefore, by employing this factor published RFs relative to PNV can be used to estimate the RF relative to 1750 ( Table 2.8 ).
In all the published studies, the RF showed a very high degree of spatial variability, with some areas showing no RF in 1990 relative to 1750 while values more negative than –5 Wm–2 were typically found in the major agricultural areas of North America and Eurasia. The local RF depends on local albedo changes, which depend on the nature of the PNV replaced by agriculture (see top panel of Figure 2.15 ). In historical simulations, the spatial patterns of RF relative to the PNV remain generally similar over time, with the regional RFs in 1750 intensifying and expanding in the area covered. The major new areas of land cover change since 1750 are North America and central and eastern Russia.
Changes in the underlying surface albedo could affect the RF due to aerosols if such changes took place in the same regions. Similarly, surface albedo RF may depend on aerosol concentrations. Estimates of the temporal evolution of aerosol RF and surface albedo RF may need to consider changes in each other ( Betts et al., 2007 [MoS, SRC, 2007] ).
Table 2.8 .Estimates of forest area, contribution to CO2 increase from anthropogenic land cover change, and RF due to the land use change-induced CO2 increase and surface albedo change, relative to pre-industrial vegetation and PNV. The CO2 RFs are for 2000 relative to 1850, calculated from the land use change contribution to the total increase in CO2 from 1850 to 2000 simulated with both land use and fossil fuel emissions by the carbon cycle models. Carbon emissions from land cover change for the 1980 s and 1990 s are discussed in Section 7.3 and Table 7.2 .
| Main Source of Land Cover Data | Forest Area PNV 106 km2 | Forest Area circa 1700 106 km2 | Forest Area circa 1990 106 km2 | Contribution to CO2 Increase 1850–2000a (ppm) | CO2 RF (Wm–2) | Albedo RF vs. PNV (Wm–2) | Albedo RF vs. 1750 (Wm–2) |
|---|---|---|---|---|---|---|---|
| Ramankutty and Foley (1999) | 55.27 | 52.77b | 43.97c | 16d | 0.27 | –0.24e | –0.18e |
| –0.29 to +0.02f | –0.22 to +0.02h | ||||||
| –0.2g | –0.14g,i | ||||||
| –0.15 to –0.28i,j | |||||||
| –0.15k | |||||||
| –0.075 to –0.325i,l | |||||||
| Klein Goldewijk (2001) | 58.6 | 54.4 | 41.5 | 12d | 0.20 | –0.66 to +0.1f | –0.50 to +0.08h |
| –0.275i,l | |||||||
| Houghton (1983m,2003) | 62.15 | 50.53n | 35d | 0.57 | |||
| 26o | 0.44 | ||||||
| MODIS (Schaaf et al., 2002) | –0.09p | –0.07h | |||||
| Wilson and Henderson-Sellers (1985) | –0.2q | –0.15h | |||||
| –0.29f | –0.22h | ||||||
| SARBr | –0.11 to –0.55f | –0.08 to –0.41h | |||||
| Matthews (1983) | –0.12f–0.4s | –0.09h–0.3h | |||||
| –0.4s | –0.3h | ||||||
| –0.08t | –0.06h |
2.5.3.1 Uncertainties
Uncertainties in estimates of RF due to anthropogenic surface albedo change arise from several factors.
2.5.3.1.1 Uncertainties in the mapping and characterisation of present-day vegetation
The RF estimates reported in the TAR used atlas-based data sets for present-day vegetation ( (Matthews, 1983; ) Wilson and Henderson-Sellers, 1985 [MoS, ARC] ). More recent data sets of land cover have been obtained from satellite remote sensing. Data from the AVHRR in 1992 to 1993 were used to generate two global land cover data sets at 1 km resolution using different methodologies ( (Hansen and Reed, 2000; ) ( Loveland et al., 2000 ) ) The International Geosphere-Biosphere Programme Data and Information System (IGBP-DIS) data set is used as the basis for global cropland maps ( Ramankutty and Foley, 1999 [JoC, MoS, SRC] ) and historical reconstructions of croplands, pasture and other vegetation types ( Ramankutty and Foley, 1999 [JoC, MoS, SRC] ; Klein Goldewijk, 2001 [JoC, MoS] )( Table 2.8 ). The MODIS ( (Friedl et al., 2002 ) ) and Global Land Cover 2000 ( (Bartholome and Belward, 2005 ) ) provide other products. The two interpretations of the AVHRR data agree on the classification of vegetation as either tall (forest and woody savannah) or short (all other land cover) over 84% of the land surface ( (Hansen and Reed, 2000 ) ). However, some of the key disagreements are in regions subject to anthropogenic land cover change so may be important for the estimation of anthropogenic RF. Using the Hadley Centre Atmospheric Model (HadAM3) GCM, Betts et al. 2007 [MoS, SRC, 2007] ) found that the estimate of RF relative to PNV varied from –0.2 Wm–2 with the Wilson and Henderson-Sellers 1985 [MoS, ARC] ) atlas-based land use data set to –0.24 Wm–2 with a version of the Wilson and Henderson-Sellers 1985 [MoS, ARC] ) data set adjusted to agree with the cropland data of Ramankutty and Foley 1999 [JoC, MoS, SRC] Myhre and Myhre 2003 [JoC, MoS, SRC] ) found the RF relative to PNV to vary from –0.66 Wm–2 to 0.29 Wm–2 according to whether the present-day land cover was from Wilson and Henderson-Sellers 1985 [MoS, ARC] Ramankutty and Foley 1999 [JoC, MoS, SRC] ) or other sources.
2.5.3.1.2 Uncertainties in the mapping and characterisation of the reference historical state
Reconstructions of historical land use states require information or assumptions regarding the nature and extent of land under human use and the nature of the PNV. Ramankutty and Foley 1999 [JoC, MoS, SRC] ) reconstructed the fraction of land under crops at 0.5° resolution from 1700 to 1990 ( Figure 2.15 , Table 2.8 )by combining the IGBP Global Land Cover Dataset with historical inventory data, assuming that all areas of past vegetation occur within areas of current vegetation. Klein Goldewijk 2001 [JoC, MoS] ) reconstructed all land cover types from 1700 to 1990 ( Figure 2.15 , Table 2.8 ), combining cropland and pasture inventory data with historical population density maps and PNV. Klein Goldewijk used a Boolean approach, which meant that crops, for example, covered either 100% or 0% of a 0.5° grid box. The total global cropland of Klein Goldewijk is generally 25% less than that reconstructed by Ramankutty and Foley 1999 [JoC, MoS, SRC] ) throughout 1700 to 1990 . At local scales, the disagreement is greater due to the high spatial heterogeneity in both data sets. Large-scale PNV( Figure 2.15 )is reconstructed either with models or by assuming that small-scale examples of currently undisturbed vegetation are representative of the PNV at the large scale. Matthews et al. 2004 [JoC, ARC] ) simulated RF relative to 1700 as –0.20 Wm–2 and –0.28 Wm–2 with the above land use reconstructions.
2.5.3.1.3 Uncertainties in the parametrizations of the surface radiation processes
The albedo for a given land surface or vegetation type may either be prescribed or simulated on the basis of more fundamental characteristics such as vegetation leaf area. But either way, model parameters are set on the basis of observational data that may come from a number of conflicting sources. Both the AVHRR and MODIS ( (Schaaf et al., 2002; ) Gao et al., 2005 [JoC, MoS] ) instruments have been used to quantify surface albedo for the IGBP vegetation classes in different regions and different seasons, and in some cases the albedo for a given vegetation type derived from one source can be twice that derived from the other (e.g., Strugnell et al., 2001 [JoC, MoS] ; Myhre et al., 2005a [JoC, MoS, SRC] Myhre and Myhre 2003 [JoC, MoS, SRC] ) examined the implications of varying the albedo of different vegetation types either together or separately, and found the RF relative to PNV to vary from –0.65 Wm–2 to +0.47 Wm–2;however, the positive RFs occurred in only a few cases and resulted from large reductions in surface albedo in semi-arid regions on conversion to pasture, so were considered unrealistic by the study’s authors. The single most important factor for the uncertainty in the study by Myhre and Myhre 2003 [JoC, MoS, SRC] ) was found to be the surface albedo for cropland. In simulations where only the cropland surface albedo was varied between 0.15, 0.18 and 0.20, the resulting RFs relative to PNV were –0.06, –0.20 and –0.29 Wm–2,respectively. Similar results were found by Matthews et al. 2003 [JoC, MoS, ARC] ) considering only cropland changes and not pasture; with cropland surface albedos of 0.17 and 0.20, RFs relative to 1700 were –0.15 and –0.28 Wm–2,respectively.
2.5.3.1.3 Uncertainties in other parts of the model
When climate models are used to estimate the RF, uncertainties in other parts of the model also affect the estimates. In particular, the simulation of snow cover affects the extent to which land cover changes affect surface albedo. Betts 2000 [JoC, SRC] ) estimated that the systematic biases in snow cover in HadAM3 introduced errors of up to approximately 10% in the simulation of local RF due to conversion between forest and open land. Such uncertainties could be reduced by the use of an observational snow climatology in a model that just treats the radiative transfer ( Myhre and Myhre, 2003 [JoC, MoS, SRC] ). The simulation of cloud cover affects the extent to which the simulated surface albedo changes affect planetary albedo – too much cloud cover could diminish the contribution of surface albedo changes to the planetary albedo change.
On the basis of the studies assessed here, including a number of new estimates since the TAR, the assessment is that the best estimate of RF relative to 1750 due to land-use related surface albedo change should remain at –0.2 ± 0.2 Wm–2.In the light of the additional modelling studies, the exclusion of feedbacks, the improved incorporation of large-scale observations and the explicit consideration of land use reconstructions for 1750, the level of scientific understanding is raised to medium-low, compared to low in the TAR( Section 2.9 , Table 2.11 ).
2.5.4 Radiative Forcing by Anthropogenic Surface Albedo Change: Black Carbon in Snow and Ice
The presence of soot particles in snow could cause a decrease in the albedo of snow and affect snowmelt. Initial estimates by Hansen et al. 2000 [JoC, ARC] ) suggested that BC could thereby exert a positive RF of +0.2 Wm–2.This estimate was refined by Hansen and Nazarenko 2004 [JoC, MoS, ARC] ), who used measured BC concentrations within snow and ice at a wide range of geographic locations to deduce the perturbation to the surface and planetary albedo, deriving an RF of +0.15 Wm–2.The uncertainty in this estimate is substantial due to uncertainties in whether BC and snow particles are internally or externally mixed, in BC and snow particle shapes and sizes, in voids within BC particles, and in the BC imaginary refractive index. Jacobson 2004 [JoC] ) developed a global model that allows the BC aerosol to enter snow via precipitation and dry deposition, thereby modifying the snow albedo and emissivity. They found modelled concentrations of BC within snow that were in reasonable agreement with those from many observations. The model study found that BC on snow and sea ice caused a decrease in the surface albedo of 0.4% globally and 1% in the NH, although RFs were not reported. Hansen et al. 2005 [JoC, MoS, ARC] ) allowed the albedo change to be proportional to local BC deposition according to Koch 2001 [JoC, MoS] ) and presented a further revised estimate of 0.08 Wm–2.They also suggested that this RF mechanism produces a greater temperature response by a factor of 1.7 than an equivalent CO2 RF, that is, the ‘efficacy’ may be higher for this RF mechanism (see Section 2.8.5.7 ). This report adopts a best estimate for the BC on snow RF of +0.10 ± 0.10 Wm–2,with a low level of scientific understanding( Section 2.9 , Table 2.11 ).
Since the dominant aspect of land cover change since 1750 has been deforestation in temperate regions, the overall effect of anthropogenic land cover change on global temperature will depend largely on the relative importance of increased surface albedo in winter and spring (exerting a cooling) and reduced evaporation in summer and in the tropics (exerting a warming) ( Bounoua et al., 2002 [JoC] ). Estimates of global temperature responses from past deforestation vary from 0.01°C ( Zhao et al., 2001 [JoC, ARC] ) to –0.25°C ( Govindasamy et al., 2001a [JoC] ; Brovkin et al., 2006 [JoC, MoS, ARC] ). If cooling by increased surface albedo dominates, then the historical effect of land cover change may still be adequately represented by RF. With tropical deforestation becoming more significant in recent decades, warming due to reduced evaporation may become more significant globally than increased surface albedo. Radiative forcing would then be less useful as a metric of climate change induced by land cover change recently and in the future.
2.5.5 Other Effects of Anthropogenic Changes in Land Cover
Anthropogenic land use and land cover change can also modify climate through other mechanisms, some directly perturbing the Earth radiation budget and some perturbing other processes. Impacts of land cover change on emissions of CO2,CH4,biomass burning aerosols and dust aerosols are discussed in Sections 2.3 and 2.4 .Land cover change itself can also modify the surface energy and moisture budgets through changes in evaporation and the fluxes of latent and sensible heat, directly affecting precipitation and atmospheric circulation as well as temperature. Model results suggest that the combined effects of past tropical deforestation may have exerted regional warmings of approximately 0.2°C relative to PNV, and may have perturbed the global atmospheric circulation affecting regional climates remote from the land cover change ( Chase et al., 2000 [JoC, MoS] ; Zhao et al., 2001 [JoC, ARC] ; Pielke et al., 2002 [JoC] ; Chapters 7 , 9 and 11 ).
2.5.6 Tropospheric Water Vapour from Anthropogenic Sources
Anthropogenic use of water is less than 1% of natural sources of water vapour and about 70% of the use of water for human activity is from irrigation ( Döll, 2002 [JoC] ). Several regional studies have indicated an impact of irrigation on temperature, humidity and precipitation ( Barnston and Schickedanz, 1984 [JoC] ; Lohar and Pal, 1995 [JoC] ; de Ridder and Gallée, 1998 [JoC] ; Moore and Rojstaczer, 2001 [JoC] ; Zhang et al., 2002 [ARC] Boucher et al. 2004 [JoC, SRC] ) used a GCM to show that irrigation has a global impact on temperature and humidity. Over Asia where most of the irrigation takes place, the simulations showed a change in the water vapour content in the lower troposphere of up to 1%, resulting in an RF of +0.03 Wm–2.However, the effect of irrigation on surface temperature was dominated by evaporative cooling rather than by the excess greenhouse effect and thus a decrease in surface temperature was found. Irrigation affects the temperature, humidity, clouds and precipitation as well as the natural evaporation through changes in the surface temperature, raising questions about the strict use of RF in this case. Uncertainties in the water vapour flow to the atmosphere from irrigation are significant and Gordon et al. 2005 [JoC] ) gave a substantially higher estimate compared to that of Boucher et al. 2004 [JoC, SRC] ). Most of this uncertainty is likely to be linked to differences between the total withdrawal for irrigation and the amount actually used ( Boucher et al., 2004 [JoC, SRC] ). Furthermore, Gordon et al. 2005 [JoC] ) also estimated a reduced water vapour flow to the atmosphere from deforestation, most importantly in tropical areas. This reduced water vapour flow is a factor of three larger than the water vapour increase due to irrigation in Boucher et al. 2004 [JoC, SRC] ), but so far there are no estimates of the effect of this on the water vapour content of the atmosphere and its RF. Water vapour changes from deforestation will, like irrigation, affect the surface evaporation and temperature and the water cycle in the atmosphere. Radiative forcing from anthropogenic sources of tropospheric water vapour is not evaluated here, since these sources affect surface temperature more significantly through these non-radiative processes, and a strict use of the RF is problematic. The emission of water vapour from fossil fuel combustion is significantly lower than the emission from changes in land use ( Boucher et al., 2004 [JoC, SRC] ).
Urban heat islands result partly from the physical properties of the urban landscape and partly from the release of heat into the environment by the use of energy for human activities such as heating buildings and powering appliances and vehicles (‘human energy production’). The global total heat flux from this is estimated as 0.03 Wm–2 ( Nakicenovic, 1998 [NPR] ). If this energy release were concentrated in cities, which are estimated to cover 0.046% of the Earth’s surface ( (Loveland et al., 2000 ) ) the mean local heat flux in a city would be 65 Wm–2.Daytime values in central Tokyo typically exceed 400 Wm–2 with a maximum of 1,590 Wm–2 in winter ( (Ichinose et al., 1999 ) ). Although human energy production is a small influence at the global scale, it may be very important for climate changes in cities ( Betts and Best, 2004 [NPR, MoS, SRC] ;( Crutzen, 2004 ) ).
2.5.8 Effects of Carbon Dioxide Changes on Climate via Plant Physiology: ‘Physiological Forcing’
As well as exerting an RF on the climate system, increasing concentrations of atmospheric CO2 can perturb the climate system through direct effects on plant physiology. Plant stomatal apertures open less under higher CO2 concentrations ( (Field et al., 1995 ) ), which directly reduces the flux of moisture from the surface to the atmosphere through transpiration ( Sellers et al., 1996 [JoC] ). A decrease in moisture flux modifies the surface energy balance, increasing the ratio of sensible heat flux to latent heat flux and therefore warming the air near the surface ( Sellers et al., 1996 [JoC] ; Betts et al., 1997 [JoC, MoS, SRC] ; Cox et al., 1999 [JoC, MoS, ARC] Betts et al. 2004 [MoS, SRC] ) proposed the term ‘physiological forcing’ for this mechanism. Although no studies have yet explicitly quantified the present-day temperature response to physiological forcing, the presence of this forcing has been detected in global hydrological budgets ( Gedney et al., 2006 [JoC] ; Section 9.5 ). This process can be considered a non-initial radiative effect, as distinct from a feedback, since the mechanism involves a direct response to increasing atmospheric CO2 and not a response to climate change. It is not possible to quantify this with RF. Reduced global transpiration would also be expected to reduce atmospheric water vapour causing a negative forcing, but no estimates of this have been made.
Increased CO2 concentrations can also ‘fertilize’ plants by stimulating photosynthesis, which models suggest has contributed to increased vegetation cover and leaf area over the 20th century ( Cramer et al., 2001 [MoS] ). Increases in the Normalized Difference Vegetation Index, a remote sensing product indicative of leaf area, biomass and potential photosynthesis, have been observed ( Zhou et al., 2001 [JoC, ARC] ), although other causes including climate change itself are also likely to have contributed. Increased vegetation cover and leaf area would decrease surface albedo, which would act to oppose the increase in albedo due to deforestation. The RF due to this process has not been evaluated and there isa very low scientific understanding of these effects.
2.6 Contrails and Aircraft-Induced Cloudiness
2.6.1 Introduction
The IPCC separately evaluated the RF from subsonic and supersonic aircraft operations in the Special Report on Aviation and the Global Atmosphere ( IPCC, 1999 [NPR] ), hereinafter designated as IPCC- 1999 . Like many other sectors, subsonic aircraft operations around the globe contribute directly and indirectly to the RF of climate change. This section only assesses the aspects that are unique to the aviation sector, namely the formation of persistent condensation trails (contrails), their impact on cirrus cloudiness, and the effects of aviation aerosols. Persistent contrail formation and induced cloudiness are indirect effects from aircraft operations because they depend on variable humidity and temperature conditions along aircraft flight tracks. Thus, future changes in atmospheric humidity and temperature distributions in the upper troposphere will have consequences for aviation-induced cloudiness. Also noted here is the potential role of aviation aerosols in altering the properties of clouds that form later in air containing aircraft emissions.
2.6.2 Radiative Forcing Estimates for Persistent Line-Shaped Contrails
Aircraft produce persistent contrails in the upper troposphere in ice-supersaturated air masses ( IPCC, 1999 [NPR] ). Contrails are thin cirrus clouds, which reflect solar radiation and trap outgoing longwave radiation. The latter effect is expected to dominate for thin cirrus ( Hartmann et al., 1992 [JoC] ; Meerkötter et al., 1999 [MoS] ), thereby resulting in a net positive RF value for contrails. Persistent contrail cover has been calculated globally from meteorological data (e.g., Sausen et al., 1998 [SRC] ) or by using a modified cirrus cloud parametrization in a GCM ( Ponater et al., 2002 [JoC, MoS, SRC] ). Contrail cover calculations are uncertain because the extent of supersaturated regions in the atmosphere is poorly known. The associated contrail RF follows from determining an optical depth for the computed contrail cover. The global RF values for contrail and induced cloudiness are assumed to vary linearly with distances flown by the global fleet if flight ambient conditions remain unchanged. The current best estimate for the RF of persistent linear contrails for aircraft operations in 2000 is +0.010 Wm–2 ( Table 2.9 ;Sausen et al., 2005 [MoS, SRC] ). The value is based on independent estimates derived from Myhre and Stordal 2001b [JoC, SRC] and Marquart et al. 2003 [JoC, MoS, SRC] ) that were updated for increased aircraft traffic in Sausen et al. 2005 [MoS, SRC] ) to give RF estimates of +0.015 Wm–2 and +0.006 Wm–2,respectively. The uncertainty range is conservatively estimated to be a factor of three. The +0.010 Wm–2 value is also considered to be the best estimate for 2005 because of the slow overall growth in aviation fuel use in the 2000 to 2005 period. The decrease in the best estimate from the TAR by a factor of two results from reassessments of persistent contrail cover and lower optical depth estimates ( Marquart and Mayer, 2002 [JoC, MoS] ; Meyer et al., 2002 [JoC, MoS] ; Ponater et al., 2002 [JoC, MoS, SRC] ; Marquart et al., 2003 [JoC, MoS, SRC] ). The new estimates include diurnal changes in the solar RF, which decreases the net RF for a given contrail cover by about 20% ( Myhre and Stordal, 2001b [JoC, SRC] ). The level of scientific understanding of contrail RF is considered low, since important uncertainties remain in the determination of global values( Section 2.9 , Table 2.11 ). For example, unexplained regional differences are found in contrail optical depths between Europe and the USA that have not been fully accounted for in model calculations ( Meyer et al., 2002 [JoC, MoS] ; Ponater et al., 2002 [JoC, MoS, SRC] ;( Palikonda et al., 2005 ) ).
Table 2.9 .Radiative forcing terms for contrail and cirrus effects caused by global subsonic aircraft operations.
| Radiative forcing (Wm–2)a | |||
|---|---|---|---|
| 1992 IPCCb | 2000 IPCCc | 2000d | |
| CO2d | 0.018 | 0.025 |
0.025 |
| Persistent linear contrails | 0.020 | 0.034 |
0.010 (0.006 to 0.015) |
| Aviation-induced cloudiness without persistent contrails | 0to 0.040 | n.a. |
|
| Aviation-induced cloudiness with persistent contrails |
0.030 (0.010 to 0.080) | ||
2.6.3 Radiative Forcing Estimates for Aviation- Induced Cloudiness
Individual persistent contrails are routinely observed to shear and spread, covering large additional areas with cirrus cloud ( Minnis et al., 1998 [JoC] ). Aviation aerosol could also lead to changes in cirrus cloud (see Section 2.6.4 ). Aviation-induced cloudiness (AIC) is defined to be the sumx of all changes in cloudiness associated with aviation operations. Thus, an AIC estimate includes persistent contrail cover. Because spreading contrails lose their characteristic linear shape, a component of AIC is indistinguishable from background cirrus. This basic ambiguity, which prevented the formulation of a best estimate of AIC amounts and the associated RF in IPCC- 1999, still exists for this assessment. Estimates of the ratio of induced cloudiness cover to that of persistent linear contrails range from 1.8 to 10 ( Minnis et al., 2004 [JoC] ;( Mannstein and Schumann, 2005 ) [10] ), indicating the uncertainty in estimating AIC amounts. Initial attempts to quantify AIC used trend differences in cirrus cloudiness between regions of high and low aviation fuel consumption ( Boucher, 1999 [JoC, SRC] ). Since IPCC- 1999, two studies have also found significant positive trends in cirrus cloudiness in some regions of high air traffic and found lower to negative trends outside air traffic regions ( (Zerefos et al., 2003; ) ( Stordal et al., 2005 ) ). Using the International Satellite Cloud Climatology Project (ISCCP) database, these studies derived cirrus cover trends for Europe of 1 to 2% per decade over the last one to two decades. A study with the Television Infrared Observation Satellite (TIROS) Operational Vertical Sounder (TOVS) provides further support for these trends ( Stubenrauch and Schumann, 2005 [JoC] ). However, cirrus trends that occurred due to natural variability, climate change or other anthropogenic effects could not be accounted for in these studies. Cirrus trends over the USA (but not over Europe) were found to be consistent with changes in contrail cover and frequency ( Minnis et al., 2004 [JoC] ). Thus, significant uncertainty remains in attributing observed cirrus trends to aviation.
Regional cirrus trends were used as a basis to compute a global mean RF value for AIC in 2000 of +0.030 Wm–2 with a range of +0.01 to +0.08 Wm–2( (Stordal et al., 2005 ) ). This value is not considered a best estimate because of the uncertainty in the optical properties of AIC and in the assumptions used to derive AIC cover. However, this value is in good agreement with the upper limit estimate for AIC RF in 1992 of +0.026 Wm–2 derived from surface and satellite cloudiness observations ( Minnis et al., 2004 [JoC] ). A value of +0.03 Wm–2 is close to the upper-limit estimate of +0.04 Wm–2 derived for non-contrail cloudiness in IPCC- 1999 . Without an AIC best estimate, the best estimate of the total RF value for aviation-induced cloudiness( Section 2.9.2 ,Table 2.12 and Figure 2.20 )includes only that due to persistent linear contrails. Radiative forcing estimates for AIC made using cirrus trend data necessarily cannot distinguish between the components of aviation cloudiness, namely persistent linear contrails, spreading contrails and other aviation aerosol effects. Some aviation effects might be more appropriately considered feedback processes rather than an RF (see Sections 2.2 and 2.4.5 ). However, the low understanding of the processes involved and the lack of quantitative approaches preclude reliably making the forcing/feedback distinction for all aviation effects in this assessment.
Two issues related to the climate response of aviation cloudiness are worth noting here. First, Minnis et al. 2004 [JoC] , 2005 ) used their RF estimate for total AIC over the USA in an empirical model, and concluded that the surface temperature response for the period 1973 to 1994 could be as large as the observed surface warming over the USA (around 0.3°C per decade). In response to the Minnis et al. conclusion, contrail RF was examined in two global climate modelling studies ( Hansen et al., 2005 [JoC, MoS, ARC] ; Ponater et al., 2005 [JoC, SRC] ). Both studies concluded that the surface temperature response calculated by Minnis et al. 2004 [JoC] ) is too large by one to two orders of magnitude. For the Minnis et al. result to be correct, the climate efficacy or climate sensitivity of contrail RF would need to be much greater than that of other larger RF terms, (e.g., CO2). Instead, contrail RF is found to have a smaller efficacy than an equivalent CO2 RF ( Hansen et al., 2005 [JoC, MoS, ARC] ; Ponater et al., 2005 [JoC, SRC] ) (see Section 2.8.5.7 ), which is consistent with the general ineffectiveness of high clouds in influencing diurnal surface temperatures ( Hansen et al., 1995 [ARC] , 2005 ). Several substantive explanations for the incorrectness of the enhanced response found in the Minnis et al. study have been presented ( Hansen et al., 2005 [JoC, MoS, ARC] ; Ponater et al., 2005 [JoC, SRC] ; Shine, 2005 [JoC] ).
The second issue is that the absence of AIC has been proposed as the cause of the increased diurnal temperature range (DTR) found in surface observations made during the short period when all USA air traffic was grounded starting on 11 September 2001 ( Travis et al., 2002 [JoC] , 2004 ). The Travis et al. studies show that during this period: (i) DTR was enhanced across the conterminous USA, with increases in the maximum temperatures that were not matched by increases of similar magnitude in the minimum temperatures, and (ii) the largest DTR changes corresponded to regions with the greatest contrail cover. The Travis et al. conclusions are weak because they are based on a correlation rather than a quantitative model and rely (necessarily) on very limited data ( (Schumann, 2005 ) ). Unusually clear weather across the USA during the shutdown period also has been proposed to account for the observed DTR changes ( (Kalkstein and Balling, 2004 ) ). Thus, more evidence and a quantitative physical model are needed before the validity of the proposed relationship between regional contrail cover and DTR can be considered further.
2.6.4 Aviation Aerosols
Global aviation operations emit aerosols and aerosol precursors into the upper troposphere and lower stratosphere ( IPCC, 1999 [NPR] ; Hendricks et al., 2004 [MoS] ). As a result, aerosol number and/or mass are enhanced above background values in these regions. Aviation-induced cloudiness includes the possible influence of aviation aerosol on cirrus cloudiness amounts. The most important aerosols are those composed of sulphate and BC (soot). Sulphate aerosols arise from the emissions of fuel sulphur and BC aerosol results from incomplete combustion of aviation fuel. Aviation operations cause enhancements of sulphate and BC in the background atmosphere ( IPCC, 1999 [NPR] ; Hendricks et al., 2004 [MoS] ). An important concern is that aviation aerosol can act as nuclei in ice cloud formation, thereby altering the microphysical properties of clouds ( Jensen and Toon, 1997 [JoC] ; Kärcher, 1999 [ARC] ; Lohmann et al., 2004 [JoC, SRC] ) and perhaps cloud cover. A modelling study by Hendricks et al. 2005 [JoC, SRC] ) showed the potential for significant cirrus modifications by aviation caused by increased numbers of BC particles. The modifications would occur in flight corridors as well as in regions far away from flight corridors because of aerosol transport. In the study, aviation aerosols either increase or decrease ice nuclei in background cirrus clouds, depending on assumptions about the cloud formation process. Results from a cloud chamber experiment showed that a sulphate coating on soot particles reduced their effectiveness as ice nuclei ( Möhler et al., 2005 [JoC] ). Changes in ice nuclei number or nucleation properties of aerosols can alter the radiative properties of cirrus clouds and, hence, their radiative impact on the climate system, similar to the aerosol-cloud interactions discussed in Sections 2.4.1 , 2.4.5 and 7.5 .No estimates are yet available for the global or regional RF changes caused by the effect of aviation aerosol on background cloudiness, although some of the RF from AIC, determined by correlation studies (see Section 2.6.3 ), may be associated with these aerosol effects.
2.7 Natural Forcings
2.7.1 Solar Variability
The estimates of long-term solar irradiance changes used in the TAR (e.g., Hoyt and Schatten, 1993 [JoC] ; Lean et al., 1995 [PoC, JoC, SRC] ) have been revised downwards, based on new studies indicating that bright solar faculae likely contributed a smaller irradiance increase since the Maunder Minimum than was originally suggested by the range of brightness in Sun-like stars ( (Hall and Lockwood, 2004; ) M. Wang et al., 2005 [Ambiguous] ). However, empirical results since the TAR have strengthened the evidence for solar forcing of climate change by identifying detectable tropospheric changes associated with solar variability, including during the solar cycle( Section 9.2 ;van Loon and Shea, 2000 [JoC, ARC] ; Douglass and Clader, 2002 [JoC] ; Gleisner and Thejll, 2003 [JoC] ; Haigh, 2003 [JoC, ARC] ; Stott et al., 2003 [JoC, MoS] ; White et al., 2003 [JoC, ARC] ; Coughlin and Tung, 2004 [JoC] ;( Labitzke, 2004; ) Crooks and Gray, 2005 [JoC, ARC] ). The most likely mechanism is considered to be some combination of direct forcing by changes in total solar irradiance, and indirect effects of ultraviolet (UV) radiation on the stratosphere. Least certain, and under ongoing debate as discussed in the TAR, are indirect effects induced by galactic cosmic rays (e.g., ( Marsh and Svensmark, 2000a, ) b; Kristjánsson et al., 2002 [NPR, JoC] ; Sun and Bradley, 2002 [PoC, JoC] ).
2.7.1.1 Direct Observations of Solar Irradiance
2.7.1.1.1 Satellite measurements of total solar irradiance
Four independent space-based instruments directly measure total solar irradiance at present, contributing to a database extant since November 1978 ( Fröhlich and Lean, 2004 [SRC] ). The Variability of Irradiance and Gravity Oscillations (VIRGO) experiment on the Solar Heliospheric Observatory (SOHO) has been operating since 1996, the ACRIM III on the Active Cavity Radiometer Irradiance Monitor Satellite (ACRIMSAT) since 1999 and the Earth Radiation Budget Satellite (ERBS) (intermittently) since 1984 . Most recent are the measurements made by the Total Solar Irradiance Monitor (TIM) on the Solar Radiation and Climate Experiment (SORCE) since 2003 ( (Rottman, 2005 ) ).
2.7.1.1.2 Observed decadal trends and variability
Different composite records of total solar irradiance have been constructed from different combinations of the direct radiometric measurements. The Physikalisch-Meteorologisches Observatorium Davos (PMOD) composite ( Fröhlich and Lean, 2004 [SRC] ), shown in Figure 2.16 ,combines the observations by the ACRIM I on the Solar Maximum Mission (SMM), the Hickey-Friedan radiometer on Nimbus 7, ACRIM II on the Upper Atmosphere Research Satellite (UARS) and VIRGO on SOHO by analysing the sensitivity drifts in each radiometer prior to determining radiometric offsets. In contrast, the ACRIM composite ( Willson and Mordvinov, 2003 [JoC] ), also shown in Figure 2.16 ,utilises ACRIMSAT rather than VIRGO observations in recent times and cross calibrates the reported data assuming that radiometric sensitivity drifts have already been fully accounted for. A third composite, the Space Absolute Radiometric Reference (SARR) composite, uses individual absolute irradiance measurements from the shuttle to cross calibrate satellite records ( (Dewitte et al., 2005 ) ). The gross temporal features of the composite irradiance records are very similar, each showing day-to-week variations associated with the Sun’s rotation on its axis, and decadal fluctuations arising from the 11-year solar activity cycle. But the linear slopes differ among the three different composite records, as do levels at solar activity minima ( 1986 and 1996 ). These differences are the result of different cross calibrations and drift adjustments applied to individual radiometric sensitivities when constructing the composites ( Fröhlich and Lean, 2004 [SRC] ).
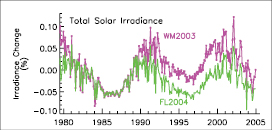
Figure 2.16. Percentage change in monthly values of the total solar irradiance composites of Willson and Mordvinov 2003 [JoC] ; WM 2003, violet symbols and line) and Fröhlich and Lean 2004 [SRC] ; FL 2004, green solid line).
Solar irradiance levels are comparable in the two most recent cycle minima when absolute uncertainties and sensitivity drifts in the measurements are assessed ( Fröhlich and Lean, 2004 [SRC] and references therein). The increase in excess of 0.04% over the 27-year period of the ACRIM irradiance composite ( Willson and Mordvinov, 2003 [JoC] ), although incompletely understood, is thought to be more of instrumental rather than solar origin ( Fröhlich and Lean, 2004 [SRC] ). The irradiance increase in the ACRIM composite is indicative of an episodic increase between 1989 and 1992 that is present in the Nimbus 7 data ( Lee et al., 1995 [JoC] ; Chapman et al., 1996 [JoC] ). Independent, overlapping ERBS observations do not show this increase; nor do they suggest a significant secular trend ( Lee et al., 1995 [JoC] ). Such a trend is not present in the PMOD composite, in which total irradiance between successive solar minima is nearly constant, to better than 0.01% ( Fröhlich and Lean, 2004 [SRC] ). Although a long-term trend of order 0.01% is present in the SARR composite between successive solar activity minima (in 1986 and 1996 ), it is not statistically significant because the estimated uncertainty is ±0.026% ( (Dewitte et al., 2005 ) ).
Current understanding of solar activity and the known sources of irradiance variability suggests comparable irradiance levels during the past two solar minima. The primary known cause of contemporary irradiance variability is the presence on the Sun’s disk of sunspots (compact, dark features where radiation is locally depleted) and faculae (extended bright features where radiation is locally enhanced). Models that combine records of the global sunspot darkening calculated directly from white light images and the magnesium (Mg) irradiance index as a proxy for the facular signal do not exhibit a significant secular trend during activity minima ( Fröhlich and Lean, 2004 [SRC] ; Preminger and Walton, 2005 [JoC, MoS] ). Nor do the modern instrumental measurements of galactic cosmic rays, 10.7 cm flux and the aa geomagnetic index since the 1950 s ( Benestad, 2005 [JoC, MoS, ARC] ) indicate this feature. While changes in surface emissivity by magnetic sunspot and facular regions are, from a theoretical view, the most effective in altering irradiance ( (Spruit, 2000 ) ), other mechanisms have also been proposed that may cause additional, possibly secular, irradiance changes. Of these, changes in solar diameter have been considered a likely candidate (e.g., Sofia and Li, 2001 [JoC] ). But recent analysis of solar imagery, primarily from the Michelson Doppler Imager (MDI) instrument on SOHO, indicates that solar diameter changes are no more than a few kilometres per year during the solar cycle ( (Dziembowski et al., 2001 ) ), for which associated irradiance changes are 0.001%, two orders of magnitude less than the measured solar irradiance cycle.
2.7.1.1.3 Measurements of solar spectral irradiance
The solar UV spectrum from 120 to 400 nm continues to be monitored from space, with SORCE observations extending those made since 1991 by two instruments on the UARS ( Woods et al., 1996 [JoC] ). SORCE also monitors, for the first time from space, solar spectral irradiance in the visible and near-infrared spectrum, providing unprecedented spectral coverage that affords a detailed characterisation of solar spectral irradiance variability. Initial results ( (Harder et al., 2005; ) Lean et al., 2005 [SRC] ) indicate that, as expected, variations occur at all wavelengths, primarily in response to changes in sunspots and faculae. Ultraviolet spectral irradiance variability in the extended database is consistent with that seen in the UARS observations since 1991, as described in the TAR.
Radiation in the visible and infrared spectrum has a notably different temporal character than the spectrum below 300 nm. Maximum energy changes occur at wavelengths from 400 to 500 nm. Fractional changes are greatest at UV wavelengths but the actual energy change is considerably smaller than in the visible spectrum. Over the time scale of the 11-year solar cycle, bolometric facular brightness exceeds sunspot blocking by about a factor of two, and there is an increase in spectral irradiance at most, if not all, wavelengths from the minimum to the maximum of the solar cycle. Estimated solar cycle changes are 0.08% in the total solar irradiance. Broken down by wavelength range these irradiance changes are 1.3% at 200 to 300 nm, 0.2% at 315 to 400 nm, 0.08% at 400 to 700 nm, 0.04% at 700 to 1,000 nm and 0.025% at 1,000 to 1,600 nm.
However, during episodes of strong solar activity, sunspot blocking can dominate facular brightening, causing decreased irradiance at most wavelengths. Spectral irradiance changes on these shorter time scales now being measured by SORCE provide tests of the wavelength-dependent sunspot and facular parametrizations in solar irradiance variability models. The modelled spectral irradiance changes are in good overall agreement with initial SORCE observations but as yet the SORCE observations are too short to provide definitive information about the amplitude of solar spectral irradiance changes during the solar cycle.
2.7.1.2 Estimating Past Solar Radiative Forcing
2.7.1.2.1 Reconstructions of past variations in solar irradiance
Long-term solar irradiance changes over the past 400 years may be less by a factor of two to four than in the reconstructions employed by the TAR for climate change simulations. Irradiance reconstructions such as those of Hoyt and Schatten 1993 [JoC] Lean et al. 1995 [PoC, JoC, SRC] Lean 2000 [JoC, SRC] Lockwood and Stamper 1999 [JoC] and Solanki and Fligge 1999 [JoC, SRC] ), used in the TAR, assumed the existence of a long-term variability component in addition to the known 11-year cycle, in which the 17th-century Maunder Minimum total irradiance was reduced in the range of 0.15% to 0.3% below contemporary solar minima. The temporal structure of this long-term component, typically associated with facular evolution, was assumed to track either the smoothed amplitude of the solar activity cycle or the cycle length. The motivation for adopting a long-term irradiance component was three-fold. Firstly, the range of variability in Sun-like stars ( Baliunas and Jastrow, 1990 [JoC] ), secondly, the long-term trend in geomagnetic activity, and thirdly, solar modulation of cosmogenic isotopes, all suggested that the Sun is capable of a broader range of activity than witnessed during recent solar cycles (i.e., the observational record in Figure 2.16 ). Various estimates of the increase in total solar irradiance from the 17th-century Maunder Minimum to the current activity minima from these irradiance reconstructions are compared with recent results in Table 2.10.
Each of the above three assumptions for the existence of a significant long-term irradiance component is now questionable. A reassessment of the stellar data was unable to recover the original bimodal separation of lower calcium (Ca) emission in non-cycling stars (assumed to be in Maunder-Minimum type states) compared with higher emission in cycling stars ( (Hall and Lockwood, 2004 ) ), which underpins the Lean et al. 1995 [PoC, JoC, SRC] and Lean 2000 [JoC, SRC] ) irradiance reconstructions. Rather, the current Sun is thought to have ‘typical’ (rather than high) activity relative to other stars. Plausible lowest brightness levels inferred from stellar observations are higher than the peak of the lower mode of the initial distribution of Baliunas and Jastrow 1990 [JoC] ). Other studies raise the possibility of long-term instrumental drifts in historical indices of geomagnetic activity ( (Svalgaard et al., 2004 ) ), which would reduce somewhat the long-term trend in the Lockwood and Stamper 1999 [JoC] ) irradiance reconstruction. Furthermore, the relationship between solar irradiance and geomagnetic and cosmogenic indices is complex, and not necessarily linear. Simulations of the transport of magnetic flux on the Sun and propagation of open flux into the heliosphere indicate that ‘open’ magnetic flux (which modulates geomagnetic activity and cosmogenic isotopes) can accumulate on inter-cycle time scales even when closed flux (such as in sunspots and faculae) does not ( Lean et al., 2002 [JoC, MoS, SRC] ; Y. Wang et al., 2005 [Ambiguous] ).
A new reconstruction of solar irradiance based on a model of solar magnetic flux variations (Y. Wang et al., 2005 [Ambiguous] ), which does not invoke geomagnetic, cosmogenic or stellar proxies, suggests that the amplitude of the background component is significantly less than previously assumed, specifically 0.27 times that of Lean 2000 [JoC, SRC] ). This estimate results from simulations of the eruption, transport and accumulation of magnetic flux during the past 300 years using a flux transport model with variable meridional flow. Variations in both the total flux and in just the flux that extends into the heliosphere (the open flux) are estimated, arising from the deposition of bipolar magnetic regions (active regions) and smaller-scale bright features (ephemeral regions) on the Sun’s surface in strengths and numbers proportional to the sunspot number. The open flux compares reasonably well with the cosmogenic isotopes for which variations arise, in part, from heliospheric modulation. This gives confidence that the approach is plausible. A small accumulation of total flux (and possibly ephemeral regions) produces a net increase in facular brightness, which, in combination with sunspot blocking, permits the reconstruction of total solar irradiance shown in Figure 2.17 .There is a 0.04% increase from the Maunder Minimum to present-day cycle minima.
Prior to direct telescopic measurements of sunspots, which commenced around 1610, knowledge of solar activity is inferred indirectly from the 14Cand 10Be cosmogenic isotope records in tree rings and ice cores, respectively, which exhibit solar-related cycles near 90, 200 and 2,300 years. Some studies of cosmogenic isotopes ( Jirikowic and Damon, 1994 [JoC] ) and spectral analysis of the sunspot record ( Rigozo et al., 2001 [MoS] ) suggest that solar activity during the 12th-century Medieval Solar Maximum was comparable to the present Modern Solar Maximum. Recent work attempts to account for the chain of physical processes in which solar magnetic fields modulate the heliosphere, in turn altering the penetration of the galactic cosmic rays, the flux of which produces the cosmogenic isotopes that are subsequently deposited in the terrestrial system following additional transport and chemical processes. An initial effort reported exceptionally high levels of solar activity in the past 70 years, relative to the preceding 8,000 years ( Solanki et al., 2004 [JoC, SRC] ). In contrast, when differences among isotopes records are taken into account and the 14Crecord corrected for fossil fuel burning, current levels of solar activity are found to be historically high, but not exceptionally so ( Muscheler et al., 2007 [JoC, ARC, 2007] ).
2.7.1.2.2 Implications for solar radiative forcing
In terms of plausible physical understanding, the most likely secular increase in total irradiance from the Maunder Minimum to current cycle minima is 0.04% (an irradiance increase of roughly 0.5 Wm–2 in 1,365 Wm–2), corresponding to an RF [11] of +0.1 Wm–2.The larger RF estimates in Table 2.10, in the range of +0.38 to +0.68 Wm–2,correspond to assumed changes in solar irradiance at cycle minima derived from brightness fluctuations in Sun-like stars that are no longer valid. Since the 11-year cycle amplitude has increased from the Maunder Minimum to the present, the total irradiance increase to the present-day cycle mean is 0.08%. From 1750 to the present there was a net 0.05% increase in total solar irradiance, according to the 11-year smoothed total solar irradiance time series of Y. Wang et al. 2005 [Ambiguous] ), shown in Figure 2.17 .This corresponds to an RF of +0.12 Wm–2,which is more than a factor of two less than the solar RF estimate in the TAR, also from 1750 to the present. Using the Lean 2000 [JoC, SRC] ) reconstruction (the lower envelope in Figure 2.17 )as an upper limit, there is a 0.12% irradiance increase since 1750, for which the RF is +0.3 Wm–2.The lower limit of the irradiance increase from 1750 to the present is 0.026% due to the increase in the 11-year cycle only. The corresponding lower limit of the RF is +0.06 Wm–2.As with solar cycle changes, long-term irradiance variations are expected to have significant spectral dependence. For example, the Y. Wang et al. 2005 [Ambiguous] ) flux transport estimates imply decreases during the Maunder Minimum relative to contemporary activity cycle minima of 0.43% at 200 to 300 nm, 0.1% at 315 to 400 nm, 0.05% at 400 to 700 nm, 0.03% at 700 to 1,000 nm and 0.02% at 1,000 to 1,600 nm ( Lean et al., 2005 [SRC] ), compared with 1.4%, 0.32%, 0.17%, 0.1% and 0.06%, respectively, in the earlier model of Lean 2000 [JoC, SRC] ).
Table 2.10. Comparison of the estimates of the increase in RF from the 17th-century Maunder Minimum (MM) to contemporary solar minima, documenting new understanding since the TAR.
| Reference | Assumptions and Technique | RF Increase from the Maunder Minimum to Contemporary Minima (Wm–2)a | Comment on Current Understanding |
|---|---|---|---|
| Schatten and Orosz (1990) | Extrapolation of the 11-year irradiance cycle to the MM, using the sunspot record. | ~0 | Irradiance levels at cycle minima remain approximately constant. |
| Lean et al. (1992) | No spots, plage or network in Ca images assumed during MM. | 0.26 | Maximum irradiance increase from a non-magnetic sun, due to changes in known bright features on contemporary solar disk. |
| Lean et al. (1992) | No spots, plage or network and reduced basal emission in cell centres in Ca images to match reduced brightness in non-cycling stars, assumed to be MM analogues. | 0.45 | New assessment of stellar data (Hall and Lockwood, 2004) does not support original stellar brightness distribution, or the use of the brightness reduction in the Baliunas and Jastrow (1990) ‘non-cycling’ stars as MM analogues. |
| Hoyt and Schatten (1993)b | Convective restructuring implied by changes in sunspot umbra/penumbra ratios from MM to present: amplitude of increase from MM to present based on brightness of non-cycling stars, from Lean et al. (1992). | 0.65 | As above |
| Lean et al. (1995) | Reduced brightness of non-cycling stars, relative to those with active cycles, assumed typical of MM. | 0.45 | As above |
| Solanki and Fligge (1999)b | Combinations of above. | 0.68 | As above |
| Lean (2000) | Reduced brightness of non-cycling stars (revised solar-stellar calibration) assumed typical of MM. | 0.38 | As above |
| Foster (2004) Model | Non-magnetic sun estimates by removing bright features from MDI images assumed for MM. | 0.28 | Similar approach to removal of spots, plage and network by Lean et al. (1992). |
| Y. Wang et al. (2005)b | Flux transport simulations of total magnetic flux evolution from MM to present. | 0.1 | Solar model suggests that modest accumulation of magnetic flux from one solar cycle to the next produces a modest increase in irradiance levels at solar cycle minima. |
| Dziembowski et al. (2001) | Helioseismic observations of solar interior oscillations suggest that the historical Sun could not have been any dimmer than current activity minima. | ~0 |
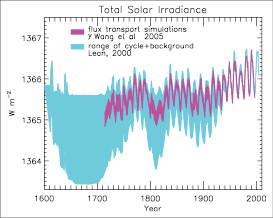
Figure 2.17. Reconstructions of the total solar irradiance time series starting as early as 1600 . The upper envelope of the shaded regions shows irradiance variations arising from the 11-year activity cycle. The lower envelope is the total irradiance reconstructed by Lean 2000 [JoC, SRC] ), in which the long-term trend was inferred from brightness changes in Sun-like stars. In comparison, the recent reconstruction of Y. Wang et al. 2005 [Ambiguous] ) is based on solar considerations alone, using a flux transport model to simulate the long-term evolution of the closed flux that generates bright faculae.
2.7.1.3 Indirect Effects of Solar Variability
Approximately 1% of the Sun’s radiant energy is in the UV portion of the spectrum at wavelengths below about 300 nm, which the Earth’s atmosphere absorbs. Although of considerably smaller absolute energy than the total irradiance, solar UV radiation is fractionally more variable by at least an order of magnitude. It contributes significantly to changes in total solar irradiance (15% of the total irradiance cycle; Lean et al., 1997 [JoC, SRC] ) and creates and modifies the ozone layer, but is not considered as a direct RF because it does not reach the troposphere. Since the TAR, new studies have confirmed and advanced the plausibility of indirect effects involving the modification of the stratosphere by solar UV irradiance variations (and possibly by solar-induced variations in the overlying mesosphere and lower thermosphere), with subsequent dynamical and radiative coupling to the troposphere( Section 9.2 ). Whether solar wind fluctuations ( Boberg and Lundstedt, 2002 [JoC] ) or solar-induced heliospheric modulation of galactic cosmic rays ( (Marsh and Svensmark, 2000b ) ) also contribute indirect forcings remains ambiguous.
As in the troposphere, anthropogenic effects, internal cycles (e.g., the Quasi-Biennial Oscillation) and natural influences all affect the stratosphere. It is now well established from both empirical and model studies that solar cycle changes in UV radiation alter middle atmospheric ozone concentrations ( Fioletov et al., 2002 [JoC, MoS] ; Geller and Smyshlyaev, 2002 [JoC, MoS, SRC] ; Hood, 2003 [JoC] ), temperatures and winds ( Ramaswamy et al., 2001 [NPR, MoS, SRC] ; Labitzke et al., 2002 [MoS] ; Haigh, 2003 [JoC, ARC] ;( Labitzke, 2004; ) Crooks and Gray, 2005 [JoC, ARC] ), including the Quasi-Biennial Oscillation ( McCormack, 2003 [JoC] ; Salby and Callaghan, 2004 [JoC] ). In their recent survey of solar influences on climate, Gray et al. 2005 [NPR, ARC] ) noted that updated observational analyses have confirmed earlier 11-year cycle signals in zonally averaged stratospheric temperature, ozone and circulation with increased statistical confidence. There is a solar-cycle induced increase in global total ozone of 2 to 3% at solar cycle maximum, accompanied by temperature responses that increase with altitude, exceeding 1°C around 50 km. However, the amplitudes and geographical and altitudinal patterns of these variations are only approximately known, and are not linked in an easily discernible manner to the forcing. For example, solar forcing appears to induce a significant lower stratospheric response ( Hood, 2003 [JoC] ), which may have a dynamical origin caused by changes in temperature affecting planetary wave propagation, but it is not currently reproduced by models.
When solar activity is high, the more complex magnetic configuration of the heliosphere reduces the flux of galactic cosmic rays in the Earth’s atmosphere. Various scenarios have been proposed whereby solar-induced galactic cosmic ray fluctuations might influence climate (as surveyed by Gray et al., 2005 [NPR, ARC] Carslaw et al. 2002 [JoC] ) suggested that since the plasma produced by cosmic ray ionization in the troposphere is part of an electric circuit that extends from the Earth’s surface to the ionosphere, cosmic rays may affect thunderstorm electrification. By altering the population of CCN and hence microphysical cloud properties (droplet number and concentration), cosmic rays may also induce processes analogous to the indirect effect of tropospheric aerosols. The presence of ions, such as produced by cosmic rays, is recognised as influencing several microphysical mechanisms ( Harrison and Carslaw, 2003 [JoC] ). Aerosols may nucleate preferentially on atmospheric cluster ions. In the case of low gas-phase sulphuric acid concentrations, ion-induced nucleation may dominate over binary sulphuric acid-water nucleation. In addition, increased ion nucleation and increased scavenging rates of aerosols in turbulent regions around clouds seem likely. Because of the difficulty in tracking the influence of one particular modification brought about by ions through the long chain of complex interacting processes, quantitative estimates of galactic cosmic-ray induced changes in aerosol and cloud formation have not been reached.
Many empirical associations have been reported between globally averaged low-level cloud cover and cosmic ray fluxes (e.g., ( Marsh and Svensmark, 2000a, ) b). Hypothesised to result from changing ionization of the atmosphere from solar-modulated cosmic ray fluxes, an empirical association of cloud cover variations during 1984 to 1990 and the solar cycle remains controversial because of uncertainties about the reality of the decadal signal itself, the phasing or anti-phasing with solar activity, and its separate dependence for low, middle and high clouds. In particular, the cosmic ray time series does not correspond to global total cloud cover after 1991 or to global low-level cloud cover after 1994 ( Kristjánsson and Kristiansen, 2000 [JoC, MoS] ; Sun and Bradley, 2002 [PoC, JoC] ) without unproven de-trending ( Usoskin et al., 2004 [JoC] ). Furthermore, the correlation is significant with low-level cloud cover based only on infrared (not visible) detection. Nor do multi-decadal ( 1952 to 1997 ) time series of cloud cover from ship synoptic reports exhibit a relationship to cosmic ray flux. However, there appears to be a small but statistically significant positive correlation between cloud over the UK and galactic cosmic ray flux during 1951 to 2000 ( (Harrison and Stephenson, 2006 ) ). Contrarily, cloud cover anomalies from 1900 to 1987 over the USA do have a signal at 11 years that is anti-phased with the galactic cosmic ray flux ( Udelhofen and Cess, 2001 [JoC] ). Because the mechanisms are uncertain, the apparent relationship between solar variability and cloud cover has been interpreted to result not only from changing cosmic ray fluxes modulated by solar activity in the heliosphere ( Usoskin et al., 2004 [JoC] ) and solar-induced changes in ozone ( Udelhofen and Cess, 2001 [JoC] ), but also from sea surface temperatures altered directly by changing total solar irradiance ( Kristjánsson et al., 2002 [NPR, JoC] ) and by internal variability due to the El Niño-Southern Oscillation ( Kernthaler et al., 1999 [JoC, ARC] ). In reality, different direct and indirect physical processes (such as those described in Section 9.2 )may operate simultaneously.
The direct RF due to increase in solar irradiance is reduced from the TAR. The best estimate is +0.12 Wm–2 (90% confidence interval: +0.06 to +0.30 Wm–2). While there have been advances in the direct solar irradiance variation, there remain large uncertainties. The level of scientific understanding is elevated to low relative to TAR for solar forcing due to direct irradiance change, while declared as very low for cosmic ray influences( Section 2.9 , Table 2.11 ).
2.7.2 Explosive Volcanic Activity
2.7.2.1 Radiative Effects of Volcanic Aerosols
Volcanic sulphate aerosols are formed as a result of oxidation of the sulphur gases emitted by explosive volcanic eruptions into the stratosphere. The process of gas-to-particle conversion has an e-folding time of roughly 35 days ( Bluth et al., 1992 [JoC] ; Read et al., 1993 [JoC] ). The e-folding time (by mass) for sedimentation of sulphate aerosols is typically about 12 to 14 months ( Lambert et al., 1993 [JoC] ; Baran and Foot, 1994 [JoC] ; Barnes and Hoffman, 1997 [JoC] ;( Bluth et al., 1997 ) ). Also emitted directly during an eruption are volcanic ash particulates (siliceous material). These are particles usually larger than 2 µm that sediment out of the stratosphere fairly rapidly due to gravity (within three months or so), but could also play a role in the radiative perturbations in the immediate aftermath of an eruption. Stratospheric aerosol data incorporated for climate change simulations tends to be mostly that of the sulphates ( Sato et al., 1993 [JoC, ARC] ; Stenchikov et al., 1998 [JoC, MoS, SRC] ; Ramachandran et al., 2000 [JoC, SRC] ; Hansen et al., 2002 [JoC, MoS, ARC] ; Tett et al., 2002 [JoC, MoS, ARC] ; Ammann et al., 2003 [PoC, JoC, MoS, ARC] ). As noted in the Second Assessment Report (SAR) and the TAR, explosive volcanic events are episodic, but the stratospheric aerosols resulting from them yield substantial transitory perturbations to the radiative energy balance of the planet, with both shortwave and longwave effects sensitive to the microphysical characteristics of the aerosols (e.g., size distribution).
Long-term ground-based and balloon-borne instrumental observations have resulted in an understanding of the optical effects and microphysical evolution of volcanic aerosols ( Deshler et al., 2003 [JoC] ; Hofmann et al., 2003 [NPR] ). Important ground-based observations of aerosol characteristics from pre-satellite era spectral extinction measurements have been analysed by Stothers 2001a [JoC] ,b), but they do not provide global coverage. Global observations of stratospheric aerosol over the last 25 years have been possible owing to a number of satellite platforms, for example, TOMS and TOVS have been used to estimate SO2 loadings from volcanic eruptions ( Krueger et al., 2000 [NPR] ; Prata et al., 2003 [NPR] ). The Stratospheric Aerosol and Gas Experiment (SAGE) and Stratospheric Aerosol Measurement (SAM) projects (e.g., ( McCormick, 1987 ) ) have provided vertically resolved stratospheric aerosol spectral extinction data for over 20 years, the longest such record. This data set has significant gaps in coverage at the time of the El Chichón eruption in 1982 (the second most important in the 20th century after Mt. Pinatubo in 1991 ) and when the aerosol cloud is dense; these gaps have been partially filled by lidar measurements and field campaigns (e.g., Antuña et al., 2003 [JoC] ; Thomason and Peter, 2006 [NPR] ).
Volcanic aerosols transported in the atmosphere to polar regions are preserved in the ice sheets, thus recording the history of the Earth’s volcanism for thousands of years ( (Bigler et al., 2002; ) ( Palmer et al., 2002; ) Mosley-Thompson et al., 2003 [NPR, MoS, ARC] ). However, the atmospheric loadings obtained from ice records suffer from uncertainties due to imprecise knowledge of the latitudinal distribution of the aerosols, depositional noise that can affect the signal for an individual eruption in a single ice core, and poor constraints on aerosol microphysical properties.
The best-documented explosive volcanic event to date, by way of reliable and accurate observations, is the 1991 eruption of Mt. Pinatubo. The growth and decay of aerosols resulting from this eruption have provided a basis for modelling the RF due to explosive volcanoes. There have been no explosive and climatically significant volcanic events since Mt. Pinatubo. As pointed out in Ramaswamy et al. 2001 [NPR, MoS, SRC] ), stratospheric aerosol concentrations are now at the lowest concentrations since the satellite era and global coverage began in about 1980 . Altitude-dependent stratospheric optical observations at a few wavelengths, together with columnar optical and physical measurements, have been used to construct the time-dependent global field of stratospheric aerosol size distribution formed in the aftermath of volcanic events. The wavelength-dependent stratospheric aerosol single-scattering characteristics calculated for the solar and longwave spectrum are deployed in climate models to account for the resulting radiative (shortwave plus longwave) perturbations.
Using available satellite- and ground-based observations, Hansen et al. 2002 [JoC, MoS, ARC] ) constructed a volcanic aerosols data set for the 1850 to 1999 period ( Sato et al., 1993 [JoC, ARC] ). This has yielded zonal mean vertically resolved aerosol optical depths for visible wavelengths and column average effective radii. Stenchikov et al. 2006 [JoC, MoS, SRC] ) introduced a slight variation to this data set, employing UARS observations to modify the effective radii relative to Hansen et al. 2002 [JoC, MoS, ARC] ), thus accounting for variations with altitude. Ammann et al. 2003 [PoC, JoC, MoS, ARC] ) developed a data set of total aerosol optical depth for the period since 1890 that does not include the Krakatau eruption. The data set is based on empirical estimates of atmospheric loadings, which are then globally distributed using a simplified parametrization of atmospheric transport, and employs a fixed aerosol effective radius (0.42 µm) for calculating optical properties. The above data sets have essentially provided the bases for the volcanic aerosols implemented in virtually all of the models that have performed the 20th-century climate integrations ( Stenchikov et al., 2006 [JoC, MoS, SRC] ). Relative to Sato et al. 1993 [JoC, ARC] ), the Ammann et al. 2003 [PoC, JoC, MoS, ARC] ) estimate yields a larger value of the optical depth, by 20 to 30% in the second part of the 20th century, and by 50% for eruptions at the end of 19th and beginning of 20th century, for example, the 1902 Santa Maria eruption( Figure 2.18 ).
The global mean RF calculated using the Sato et al. 1993 [JoC, ARC] ) data yields a peak in radiative perturbation of about –3 Wm–2 for the strong (rated in terms of emitted SO2) 1860 and 1991 eruptions of Krakatau and Mt. Pinatubo, respectively. The value is reduced to about –2 Wm–2 for the relatively less intense El Chichón and Agung eruptions ( Hansen et al., 2002 [JoC, MoS, ARC] ). As expected from the arguments above, Ammann’s RF is roughly 20 to 30% larger than Sato’s RF.
Not all features of the aerosols are well quantified, and extending and improving the data sets remains an important area of research. This includes improved estimates of the aerosol size parameters ( Bingen et al., 2004 [JoC] ), a new approach for calculating aerosol optical characteristics using SAGE and UARS data ( Bauman et al., 2003 [JoC, SRC] ), and intercomparison of data from different satellites and combining them to fill gaps ( Randall et al., 2001 [JoC] ). While the aerosol characteristics are better constrained for the Mt. Pinatubo eruption, and to some extent for the El Chichón and Agung eruptions, the reliability degrades for aerosols from explosive volcanic events further back in time as there are few, if any, observational constraints on their optical depth and size evolution.
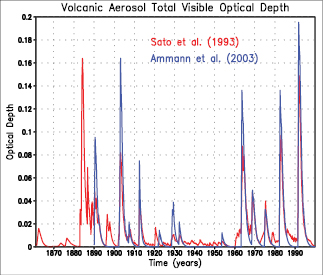
Figure 2.18. Visible (wavelength 0.55 μm) optical depth estimates of stratospheric sulphate aerosols formed in the aftermath of explosive volcanic eruptions that occurred between 1860 and 2000 . Results are shown from two different data sets that have been used in recent climate model integrations. Note that the Ammann et al. 2003 [PoC, JoC, MoS, ARC] ) data begins in 1890 .
The radiative effects due to volcanic aerosols from major eruptions are manifest in the global mean anomaly of reflected solar radiation; this variable affords a good estimate of radiative effects that can actually be tested against observations. However, unlike RF, this variable contains effects due to feedbacks (e.g., changes in cloud distributions) so that it is actually more a signature of the climate response. In the case of the Mt. Pinatubo eruption, with a peak global visible optical depth of about 0.15, simulations yield a large negative perturbation as noted above of about –3 Wm–2 ( Ramachandran et al., 2000 [JoC, SRC] ; Hansen et al., 2002 [JoC, MoS, ARC] ) (see also Section 9.2 ). This modelled estimate of reflected solar radiation compares reasonably with ERBS observations ( Minnis et al., 1993 [JoC, MoS] ). However, the ERBS observations were for a relatively short duration, and the model-observation comparisons are likely affected by differing cloud effects in simulations and measurements. It is interesting to note ( Stenchikov et al., 2006 [JoC, MoS, SRC] ) that, in the Mt. Pinatubo case, the Goddard Institute for Space Studies (GISS) models that use the Sato et al. 1993 [JoC, ARC] ) data yield an even greater solar reflection than the National Center for Atmospheric Research (NCAR) model that uses the larger ( Ammann et al., 2003 [PoC, JoC, MoS, ARC] ) optical depth estimate.
2.7.2.2 Thermal, Dynamical and Chemistry Perturbations Forced by Volcanic Aerosols
Four distinct mechanisms have been invoked with regards to the climate response to volcanic aerosol RF. First, these forcings can directly affect the Earth’s radiative balance and thus alter surface temperature. Second, they introduce horizontal and vertical heating gradients; these can alter the stratospheric circulation, in turn affecting the troposphere. Third, the forcings can interact with internal climate system variability (e.g., El Niño-Southern Oscillation, North Atlantic Oscillation, Quasi-Biennial Oscillation) and dynamical noise, thereby triggering, amplifying or shifting these modes (see Section 9.2 ;Yang and Schlesinger, 2001 [JoC] ; Stenchikov et al., 2004 [JoC, SRC] ). Fourth, volcanic aerosols provide surfaces for heterogeneous chemistry affecting global stratospheric ozone distributions ( Chipperfield et al., 2003 [NPR, MoS, SRC] ) and perturbing other trace gases for a considerable period following an eruption. Each of the above mechanisms has its own spatial and temporal response pattern. In addition, the mechanisms could depend on the background state of the climate system, and thus on other forcings (e.g., due to well-mixed gases, Meehl et al., 2004 [JoC, MoS, ARC] ), or interact with each other.
The complexity of radiative-dynamical response forced by volcanic impacts suggests that it is important to calculate aerosol radiative effects interactively within the model rather than prescribe them ( Andronova et al., 1999 [JoC, MoS, ARC] ; Broccoli et al., 2003 [JoC, MoS, ARC] ). Despite differences in volcanic aerosol parameters employed, models computing the aerosol radiative effects interactively yield tropical and global mean lower-stratospheric warmings that are fairly consistent with each other and with observations ( Ramachandran et al., 2000 [JoC, SRC] ; Hansen et al., 2002 [JoC, MoS, ARC] ; Yang and Schlesinger, 2002 [JoC] ; Stenchikov et al., 2004 [JoC, SRC] ; Ramaswamy et al., 2006b [JoC, SRC] ); however, there is a considerable range in the responses in the polar stratosphere and troposphere. The global mean warming of the lower stratosphere is due mainly to aerosol effects in the longwave spectrum, in contrast to the flux changes at the TOA that are essentially due to aerosol effects in the solar spectrum. The net radiative effects of volcanic aerosols on the thermal and hydrologic balance (e.g., surface temperature and moisture) have been highlighted by recent studies ( Free and Angell, 2002 [JoC, ARC] ; Jones et al., 2003 [NPR, PoC, ARC] ; see Chapter6 ;and see Chapter9 for significance of the simulated responses and model-observation comparisons for 20th-century eruptions). A mechanism closely linked to the optical depth perturbation and ensuing warming of the tropical lower stratosphere is the potential change in the cross-tropopause water vapour flux ( Joshi and Shine, 2003 [JoC] ; see Section 2.3.7 ).
Anomalies in the volcanic-aerosol induced global radiative heating distribution can force significant changes in atmospheric circulation, for example, perturbing the equator-to-pole heating gradient ( Stenchikov et al., 2002 [JoC, SRC] ; Ramaswamy et al., 2006a [NPR, MoS, SRC] ; see Section 9.2 )and forcing a positive phase of the Arctic Oscillation that in turn causes a counterintuitive boreal winter warming at middle and high latitudes over Eurasia and North America ( Perlwitz and Graf, 2001 [JoC] ; Stenchikov et al., 2002 [JoC, SRC] , 2004, 2006; Shindell et al., 2003b [PoC, JoC, MoS, ARC] , 2004; Perlwitz and Harnik, 2003 [JoC] ; Rind et al., 2005 [JoC, SRC] ; Miller et al., 2006 [PoC, JoC, MoS, ARC] ).
Stratospheric aerosols affect the chemistry and transport processes in the stratosphere, resulting in the depletion of ozone ( Brasseur and Granier, 1992 [JoC, ARC] ; Tie et al., 1994 [JoC, MoS, ARC] ; Solomon et al., 1996 [PoC, JoC, ARC] ; Chipperfield et al., 2003 [NPR, MoS, SRC] Stenchikov et al. 2002 [JoC, SRC] ) demonstrated a link between ozone depletion and Arctic Oscillation response; this is essentially a secondary radiative mechanism induced by volcanic aerosols through stratospheric chemistry. Stratospheric cooling in the polar region associated with a stronger polar vortex initiated by volcanic effects can increase the probability of formation of polar stratospheric clouds and therefore enhance the rate of heterogeneous chemical destruction of stratospheric ozone, especially in the NH ( Tabazadeh et al., 2002 [JoC] ). The above studies indicate effects on the stratospheric ozone layer in the wake of a volcanic eruption and under conditions of enhanced anthropogenic halogen loading. Interactive microphysics-chemistry-climate models ( Rozanov et al., 2002 [JoC, MoS] , 2004; Shindell et al., 2003b [PoC, JoC, MoS, ARC] ; Timmreck et al., 2003 [NPR] ; Dameris et al., 2005 [MoS, ARC] ) indicate that aerosol-induced stratospheric heating affects the dispersion of the volcanic aerosol cloud, thus affecting the spatial RF. However the models’ simplified treatment of aerosol microphysics introduces biases; further, they usually overestimate the mixing at the tropopause level and intensity of meridional transport in the stratosphere ( Douglass et al., 2003 [JoC, MoS] ; Schoeberl et al., 2003 [JoC, MoS] ). For present climate studies, it is practical to utilise simpler approaches that are reliably constrained by aerosol observations.
Because of its episodic and transitory nature, it is difficult to give a best estimate for the volcanic RF, unlike the other agents. Neither a best estimate nor a level of scientific understanding was given in the TAR. For the well-documented case of the explosive 1991 Mt. Pinatubo eruption, there is a good scientific understanding. However, the limited knowledge of the RF associated with prior episodic, explosive events indicatesa low level of scientific understanding( Section 2.9 , Table 2.11 ).
2.8 Utility of Radiative Forcing
The TAR and other assessments have concluded that RF is a useful tool for estimating, to a first order, the relative global climate impacts of differing climate change mechanisms ( Ramaswamy et al., 2001 [NPR, MoS, SRC] ; Jacob et al., 2005 [NPR, MoS, ARC] ). In particular, RF can be used to estimate the relative equilibrium globally averaged surface temperature change due to different forcing agents. However, RF is not a measure of other aspects of climate change or the role of emissions (see Sections 2.2 and 2.10 ). Previous GCM studies have indicated that the climate sensitivity parameter was more or less constant (varying by less than 25%) between mechanisms ( Ramaswamy et al., 2001 [NPR, MoS, SRC] ; Chipperfield et al., 2003 [NPR, MoS, SRC] ). However, this level of agreement was found not to hold for certain mechanisms such as ozone changes at some altitudes and changes in absorbing aerosol. Because the climate responses, and in particular the equilibrium climate sensitivities, exhibited by GCMs vary by much more than 25% (see Section 9.6 Ramaswamy et al. 2001 [NPR, MoS, SRC] and Jacob et al. 2005 [NPR, MoS, ARC] ) concluded that RF is the most simple and straightforward measure for the quantitative assessment of climate change mechanisms, especially for the LLGHGs. This section discusses the several studies since the TAR that have examined the relationship between RF and climate response. Note that this assessment is entirely based on climate model simulations.
2.8.1 Vertical Forcing Patterns and Surface Energy Balance Changes
The vertical structure of a forcing agent is important both for efficacy (see Section 2.8.5 )and for other aspects of climate response, particularly for evaluating regional and vertical patterns of temperature change and also changes in the hydrological cycle. For example, for absorbing aerosol, the surface forcings are arguably a more useful measure of the climate response (particularly for the hydrological cycle) than the RF ( Ramanathan et al., 2001a [JoC, SRC] ; Menon et al., 2002b [JoC, ARC] ). It should be noted that a perturbation to the surface energy budget involves sensible and latent heat fluxes besides solar and longwave irradiance; therefore, it can quantitatively be very different from the RF, which is calculated at the tropopause, and thus is not representative of the energy balance perturbation to the surface-troposphere (climate) system. While the surface forcing adds to the overall description of the total perturbation brought about by an agent, the RF and surface forcing should not be directly compared nor should the surface forcing be considered in isolation for evaluating the climate response (see, e.g., the caveats expressed in Manabe and Wetherald, 1967 [JoC] ; Ramanathan, 1981 [JoC, SRC] ). Therefore, surface forcings are presented as an important and useful diagnostic tool that aids understanding of the climate response (see Sections 2.9.4 and 2.9.5 ).
2.8.2 Spatial Patterns of Radiative Forcing
Each RF agent has a unique spatial pattern (see, e.g., Figure 6.7 in Ramaswamy et al., 2001 [NPR, MoS, SRC] ). When combining RF agents it is not just the global mean RF that needs to be considered. For example, even with a net global mean RF of zero, significant regional RFs can be present and these can affect the global mean temperature response (see Section 2.8.5 ). Spatial patterns of RF also affect the pattern of climate response. However, note that, to first order, very different RF patterns can have similar patterns of surface temperature response and the location of maximum RF is rarely coincident with the location of maximum response ( Boer and Yu, 2003b [JoC] ). Identification of different patterns of response is particularly important for attributing past climate change to particular mechanisms, and is also important for the prediction of regional patterns of future climate change. This chapter employs RF as the method for ranking the effect of a forcing agent on the equilibrium global temperature change, and only this aspect of the forcing-response relationship is discussed. However, patterns of RF are presented as a diagnostic in Section 2.9.5 .
RFs are increasingly being diagnosed from GCM integrations where the calculations are complex ( Stuber et al., 2001b [MoS, SRC] ; Tett et al., 2002 [JoC, MoS, ARC] ; Gregory et al., 2004 [JoC, MoS, ARC] ). This chapter also discusses several mechanisms that include some response in the troposphere, such as cloud changes. These mechanisms are not initially radiative in nature, but will eventually lead to a radiative perturbation of the surface-troposphere system that could conceivably be measured at the TOA. Jacob et al. 2005 [NPR, MoS, ARC] ) refer to these mechanisms as non-radiative forcings (see also Section 2.2 ). Alternatives to the standard stratospherically adjusted RF definition have been proposed that may help account for these processes. Since the TAR, several studies have employed GCMs to diagnose the zero-surface-temperature-change RF (see Figure 2.2 and Section 2.2 ). These studies have used a number of different methodologies. Shine et al. 2003 [JoC, MoS] ) fixed both land and sea surface temperatures globally and calculated a radiative energy imbalance: this technique is only feasible in GCMs with relatively simple land surface parametrizations. Hansen et al. 2005 [JoC, MoS, ARC] ) fixed sea surface temperatures and calculated an RF by adding an extra term to the radiative imbalance that took into account how much the land surface temperatures had responded. Sokolov 2006 [JoC, MoS, ARC] ) diagnosed the zero-surface-temperature-change RF by computing surface-only and atmospheric-only components of climate feedback separately in a slab model and then modifying the stratospherically adjusted RF by the atmospheric-only feedback component. Gregory et al. 2004 [JoC, MoS, ARC] ; see also Hansen et al., 2005 [JoC, MoS, ARC] ; Forster and Taylor, 2006 [JoC, MoS, SRC] ) used a regression method with a globally averaged temperature change ordinate to diagnose the zero-surface-temperature-change RF: this method had the largest uncertainties. Shine et al. 2003 [JoC, MoS] Hansen et al. 2005 [JoC, MoS, ARC] and Sokolov 2006 [JoC, MoS, ARC] ) all found that that the fixed-surface-temperature RF was a better predictor of the equilibrium global mean surface temperature response than the stratospherically adjusted RF. Further, it was a particularly useful diagnostic for changes in absorbing aerosol where the stratospherically adjusted RF could fail as a predictor of the surface temperature response (see Section 2.8.5.5 ). Differences between the zero-surface-temperature-change RF and the stratospherically adjusted RF can be caused by semi-direct and cloud-aerosol interaction effects beyond the cloud albedo RF. For most mechanisms, aside from the case of certain aerosol changes, the difference is likely to be small ( Shine et al., 2003 [JoC, MoS] ; Hansen et al., 2005 [JoC, MoS, ARC] ; Sokolov, 2006 [JoC, MoS, ARC] ). These calculations also remove problems associated with defining the tropopause in the stratospherically adjusted RF definition ( Shine et al., 2003 [JoC, MoS] ; Hansen et al., 2005 [JoC, MoS, ARC] ). However, stratospherically adjusted RF has the advantage that it does not depend on relatively uncertain components of a GCM’s response, such as cloud changes. For the LLGHGs, the stratospherically adjusted RF also has the advantage that it is readily calculated in detailed off-line radiation codes. For these reasons, the stratospherically adjusted RF is retained as the measure of comparison used in this chapter (see Section 2.2 ). However, to first order, all methods are comparable and all prove useful for understanding climate response.
2.8.4 Linearity of the Forcing-Response Relationship
Reporting findings from several studies, the TAR concluded that responses to individual RFs could be linearly added to gauge the global mean response, but not necessarily the regional response ( Ramaswamy et al., 2001 [NPR, MoS, SRC] ). Since then, studies with several equilibrium and/or transient integrations of several different GCMs have found no evidence of any nonlinearity for changes in greenhouse gases and sulphate aerosol ( Boer and Yu, 2003b [JoC] ; Gillett et al., 2004 [JoC, MoS, ARC] ; Matthews et al., 2004 [JoC, ARC] ; Meehl et al., 2004 [JoC, MoS, ARC] ). Two of these studies also examined realistic changes in many other forcing agents without finding evidence of a nonlinear response ( Meehl et al., 2004 [JoC, MoS, ARC] ; Matthews et al., 2004 [JoC, ARC] ). In all four studies, even the regional changes typically added linearly. However, Meehl et al. 2004 [JoC, MoS, ARC] ) observed that neither precipitation changes nor all regional temperature changes were linearly additive. This linear relationship also breaks down for global mean temperatures when aerosol-cloud interactions beyond the cloud albedo RF are included in GCMs ( Feichter et al., 2004 [JoC, MoS, SRC] ; see also Rotstayn and Penner, 2001 [JoC, MoS, ARC] ; Lohmann and Feichter, 2005 [SRC] ). Studies that include these effects modify clouds in their models, producing an additional radiative imbalance. Rotstayn and Penner 2001 [JoC, MoS, ARC] ) found that if these aerosol-cloud effects are accounted for as additional forcing terms, the inference of linearity can be restored (see Sections 2.8.3 and 2.8.5 ). Studies also find nonlinearities for large negative RFs, where static stability changes in the upper troposphere affect the climate feedback (e.g., Hansen et al., 2005 [JoC, MoS, ARC] ). For the magnitude and range of realistic RFs discussed in this chapter, and excluding cloud-aerosol interaction effects, there is high confidence in a linear relationship between global mean RF and global mean surface temperature response.
2.8.5 Efficacy and Effective Radiative Forcing
Efficacy (E) is defined as the ratio of the climate sensitivity parameter for a given forcing agent (λi)to the climate sensitivity parameter for CO2 changes, that is,Ei = λi / λCO2 ( Joshi et al., 2003 [JoC, MoS] ; Hansen and Nazarenko, 2004 [JoC, MoS, ARC] ). Efficacy can then be used to define an effective RF (=Ei RFi )(Joshi et al., 2003 [JoC, MoS] ; Hansen et al., 2005 [JoC, MoS, ARC] ). For the effective RF, the climate sensitivity parameter is independent of the mechanism, so comparing this forcing is equivalent to comparing the equilibrium global mean surface temperature change. That is, ΔTs =λCO2 xEi x RFi Preliminary studies have found that efficacy values for a number of forcing agents show less model dependency than the climate sensitivity values ( Joshi et al., 2003 [JoC, MoS] ). Effective RFs have been used get one step closer to an estimator of the likely surface temperature response than can be achieved by using RF alone ( Sausen and Schumann, 2000 [JoC, MoS, SRC] ; Hansen et al., 2005 [JoC, MoS, ARC] ; Lohmann and Feichter, 2005 [SRC] ). Adopting the zero-surface-temperature-change RF, which has efficacies closer to unity, may be another way of achieving similar goals (see Section 2.8.3 ). This section assesses the efficacy associated with stratospherically adjusted RF, as this is the definition of RF adopted in this chapter (see Section 2.2 ). Therefore, cloud-aerosol interaction effects beyond the cloud albedo RF are included in the efficacy term. The findings presented in this section are from an assessment of all the studies referenced in the caption of Figure 2.19 ,which presents a synthesis of efficacy results. As space is limited not all these studies are explicitly discussed in the main text.
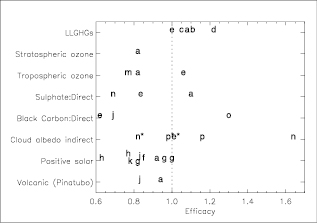
Figure 2.19. Efficacies as calculated by several GCM models for realistic changes in RF agents. Letters are centred on efficacy value and refer to the literature study that the value is taken from (see text of Section 2.8.5 for details and further discussion). In each RF category, only one result is taken per model or model formulation. Cloud-albedo efficacies are evaluated in two ways: the standard letters include cloud lifetime effects in the efficacy term and the letters with asterisks exclude these effects. Studies assessed in the figure are: a) Hansen et al. 2005 [JoC, MoS, ARC] ); b) Wang et al. 1991 [JoC, MoS] ); c) Wang et al. 1992 [JoC] ); d) Govindasamy et al. 2001b [JoC, MoS] ); e) Lohmann and Feichter 2005 [SRC] ); f) Forster et al. 2000 [JoC, MoS, SRC] ); g) Joshi et al. 2003 [JoC, MoS] ; see also Stuber et al., 2001a [JoC, SRC] ); h) Gregory et al. 2004 [JoC, MoS, ARC] ); j) Sokolov 2006 [JoC, MoS, ARC] ); k) Cook and Highwood 2004 [JoC, MoS] ); m) Mickley et al. 2004 [JoC, MoS, SRC] ); n) Rotstayn and Penner 2001 [JoC, MoS, ARC] ); o) Roberts and Jones 2004 [JoC, ARC] ) and p) Williams et al. 2001a [JoC, MoS, ARC] ).
2.8.5.1 Generic Understanding
Since the TAR, several GCM studies have calculated efficacies and a general understanding is beginning to emerge as to how and why efficacies vary between mechanisms. The initial climate state, and the sign and magnitude of the RF have less importance but can still affect efficacy ( Boer and Yu, 2003a [JoC] ; Joshi et al., 2003 [JoC, MoS] ; Hansen et al., 2005 [JoC, MoS, ARC] ). These studies have also developed useful conceptual models to help explain variations in efficacy with forcing mechanism. The efficacy primarily depends on the spatial structure of the forcings and the way they project onto the various different feedback mechanisms ( Boer and Yu, 2003b [JoC] ). Therefore, different patterns of RF and any nonlinearities in the forcing response relationship affects the efficacy ( Boer and Yu, 2003b [JoC] ; Joshi et al., 2003 [JoC, MoS] ; Hansen et al., 2005 [JoC, MoS, ARC] ; Stuber et al., 2005 [JoC, MoS, SRC] ; Sokolov, 2006 [JoC, MoS, ARC] ). Many of the studies presented in Figure 2.19 find that both the geographical and vertical distribution of the forcing can have the most significant effect on efficacy (in particular see Boer and Yu, 2003b [JoC] ; Joshi et al., 2003 [JoC, MoS] ; Stuber et al., 2005 [JoC, MoS, SRC] ; Sokolov, 2006 [JoC, MoS, ARC] ). Nearly all studies that examine it find that high-latitude forcings have higher efficacies than tropical forcings. Efficacy has also been shown to vary with the vertical distribution of an applied forcing ( Hansen et al., 1997 [JoC, MoS, ARC] ; Christiansen, 1999 [JoC, MoS] ; Joshi et al., 2003 [JoC, MoS] ; Cook and Highwood, 2004 [JoC, MoS] ; Roberts and Jones, 2004 [JoC, ARC] ; Forster and Joshi, 2005 [JoC, SRC] ; Stuber et al., 2005 [JoC, MoS, SRC] ; Sokolov, 2006 [JoC, MoS, ARC] ). Forcings that predominately affect the upper troposphere are often found to have smaller efficacies compared to those that affect the surface. However, this is not ubiquitous as climate feedbacks (such as cloud and water vapour) will depend on the static stability of the troposphere and hence the sign of the temperature change in the upper troposphere ( Govindasamy et al., 2001b [JoC, MoS] ; Joshi et al., 2003 [JoC, MoS] ; Sokolov, 2006 [JoC, MoS, ARC] ).
The few models that have examined efficacy for combined LLGHG changes generally find efficacies slightly higher than 1.0( Figure 2.19 ). Further, the most recent result from the NCAR Community Climate Model (CCM3) GCM ( Govindasamy et al., 2001b [JoC, MoS] ) indicates an efficacy of over 1.2 with no clear reason of why this changed from earlier versions of the same model. Individual LLGHG efficacies have only been analysed in two or three models. Two GCMs suggest higher efficacies from individual components (over 30% for CFCs in Hansen et al., 2005 [JoC, MoS, ARC] ). In contrast another GCM gives efficacies for CFCs ( Forster and Joshi, 2005 [JoC, SRC] ) and CH4 ( Berntsen et al., 2005 [JoC, SRC] ) that are slightly less than one. Overall there is medium confidence that the observed changes in the combined LLGHG changes have an efficacy close to 1.0 (within 10%), but there are not enough studies to constrain the efficacies for individual species.
2.8.5.3 Solar
Solar changes, compared to CO2,have less high-latitude RF and more of the RF realised at the surface. Established but incomplete knowledge suggests that there is partial compensation between these effects, at least in some models, which leads to solar efficacies close to 1.0. All models with a positive solar RF find efficacies of 1.0 or smaller. One study finds a smaller efficacy than other models (0.63: Gregory et al., 2004 [JoC, MoS, ARC] ). However, their unique methodology for calculating climate sensitivity has large uncertainties (see Section 2.8.4 ). These studies have only examined solar RF from total solar irradiance change; any indirect solar effects (see Section 2.7.1.3 )are not included in this efficacy estimate. Overall, there is medium confidence that the direct solar efficacy is within the 0.7 to 1.0 range.
Stratospheric ozone efficacies have normally been calculated from idealised ozone increases. Experiments with three models ( Stuber et al., 2001a [JoC, SRC] ; Joshi et al., 2003 [JoC, MoS] ; Stuber et al., 2005 [JoC, MoS, SRC] ) found higher efficacies for such changes; these were due to larger than otherwise tropical tropopause temperature changes which led to a positive stratospheric water vapour feedback. However, this mechanism may not operate in the two versions of the GISS model, which found smaller efficacies. Only one study has used realistic stratospheric ozone changes (see Figure 2.19 ); thus, knowledge is still incomplete. Conclusions are only drawn from the idealised studies where there is (1) medium confidence that the efficacy is within a 0.5 to 2.0 range and (2) established but incomplete physical understanding of how and why the efficacy could be larger than 1.0. There is medium confidence that for realistic tropospheric ozone perturbations the efficacy is within the 0.6 to 1.1 range.
2.8.5.5 Scattering Aerosol
For idealised global perturbations, the efficacy for the direct effect of scattering aerosol is very similar to that for changes in the solar constant ( Cook and Highwood, 2004 [JoC, MoS] ). As for ozone, realistic perturbations of scattering aerosol exhibit larger changes at higher latitudes and thus have a higher efficacy than solar changes ( Hansen et al., 2005 [JoC, MoS, ARC] ). Although the number of modelling results is limited, it is expected that efficacies would be similar to other solar effects; thus there is medium confidence that efficacies for scattering aerosol would be in the 0.7 to 1.1 range. Efficacies are likely to be similar for scattering aerosol in the troposphere and stratosphere.
With the formulation of RF employed in this chapter, the efficacy of the cloud albedo RF accounts for cloud lifetime effects( Section 2.8.3 ). Only two studies contained enough information to calculate efficacy in this way and both found efficacies higher than 1.0. However, the uncertainties in quantifying the cloud lifetime effect make this efficacy very uncertain. If cloud lifetime effects were excluded from the efficacy term, the cloud albedo efficacy would very likely be similar to that of the direct effect (see Figure 2.19 ).
2.8.5.6 Absorbing Aerosol
For absorbing aerosols, the simple ideas of a linear forcing-response relationship and efficacy can break down ( Hansen et al., 1997 [JoC, MoS, ARC] ; Cook and Highwood, 2004 [JoC, MoS] ; Feichter et al., 2004 [JoC, MoS, SRC] ; Roberts and Jones, 2004 [JoC, ARC] ; Hansen et al., 2005 [JoC, MoS, ARC] ; Penner et al., 2007 [NPR, ARC, 2007] ). Aerosols within a particular range of single scattering albedos have negative RFs but induce a global mean warming, that is, the efficacy can be negative. The surface albedo and height of the aerosol layer relative to the cloud also affects this relationship( Section 7.5 ;Penner et al., 2003 [JoC, ARC] ; Cook and Highwood, 2004 [JoC, MoS] ; Feichter et al., 2004 [JoC, MoS, SRC] ; Johnson et al., 2004 [JoC] ; Roberts and Jones, 2004 [JoC, ARC] ; Hansen et al., 2005 [JoC, MoS, ARC] ). Studies that increase BC in the planetary boundary layer find efficacies much larger than 1.0 ( Cook and Highwood, 2004 [JoC, MoS] ; Roberts and Jones, 2004 [JoC, ARC] ; Hansen et al., 2005 [JoC, MoS, ARC] ). These studies also find that efficacies are considerably smaller than 1.0 when BC aerosol is changed above the boundary layer. These changes in efficacy are at least partly attributable to a semi-direct effect whereby absorbing aerosol modifies the background temperature profile and tropospheric cloud (see Section 7.5 ). Another possible feedback mechanism is the modification of snow albedo by BC aerosol ( Hansen and Nazarenko, 2004 [JoC, MoS, ARC] ; Hansen et al., 2005 [JoC, MoS, ARC] ); however, this report does not classify this as part of the response, but rather as a separate RF (see Section 2.5.4 and 2.8.5.7). Most GCMs likely have some representation of the semi-direct effect ( Cook and Highwood, 2004 [JoC, MoS] ) but its magnitude is very uncertain (see Section 7.5 )and dependent on aspects of cloud parametrizations within GCMs ( Johnson, 2005 [JoC, MoS] ). Two studies using realistic vertical and horizontal distributions of BC find that overall the efficacy is around 0.7 ( Hansen et al., 2005 [JoC, MoS, ARC] ; Lohmann and Feichter, 2005 [SRC] ). However, Hansen et al. 2005 [JoC, MoS, ARC] ) acknowledge that they may have underestimated BC within the boundary layer and another study with realistic vertical distribution of BC changes finds an efficacy of 1.3 ( Sokolov, 2006 [JoC, MoS, ARC] ). Further, Penner et al. 2007 [NPR, ARC, 2007] ) also modelled BC changes and found efficacies very much larger and very much smaller than 1.0 for biomass and fossil fuel carbon, respectively ( Hansen et al. 2005 [JoC, MoS, ARC] ) found similar efficacies for biomass and fossil fuel carbon). In summary there is no consensus as to BC efficacy and this may represent problems with the stratospherically adjusted definition of RF (see Section 2.8.3 ).
Efficacies for some other effects have been evaluated by one or two modelling groups. Hansen et al. 2005 [JoC, MoS, ARC] ) found that land use albedo RF had an efficacy of roughly 1.0, while the BC-snow albedo RF had an efficacy of 1.7. Ponater et al. 2005 [JoC, SRC] ) found an efficacy of 0.6 for contrail RF and this agrees with a suggestion from Hansen et al. 2005 [JoC, MoS, ARC] ) that high-cloud changes should have smaller efficacies. The results of Hansen et al. 2005 [JoC, MoS, ARC] and Forster and Shine 1999 [JoC, SRC] ) suggest that stratospheric water vapour efficacies are roughly one.
2.8.6 Efficacy and the Forcing-Response Relationship
Efficacy is a new concept introduced since the TAR and its physical understanding is becoming established (see Section 2.8.5 ). When employing the stratospherically adjusted RF, there is medium confidence that efficacies are within the 0.75 to 1.25 range for most realistic RF mechanisms aside from aerosol and stratospheric ozone changes. There is medium confidence that realistic aerosol and ozone changes have efficacies within the 0.5 to 2.0 range. Further, zero-surface-temperature-change RFs are very likely to have efficacies significantly closer to 1.0 for all mechanisms. It should be noted that efficacies have only been evaluated in GCMs and actual climate efficacies could be different from those quoted in Section 2.8.5 .
2.9 Synthesis
This section begins by synthesizing the discussion of the RF concept. It presents summaries of the global mean RFs assessed in earlier sections and discusses time evolution and spatial patterns of RF. It also presents a brief synthesis of surface forcing diagnostics. It breaks down the analysis of RF in several ways to aid and advance the understanding of the drivers of climate change.
RFs are calculated in various ways depending on the agent: from changes in emissions and/or changes in concentrations; and from observations and other knowledge of climate change drivers. Current RF depends on present-day concentrations of a forcing agent, which in turn depend on the past history of emissions. Some climate response to these RFs is expected to have already occurred. Additionally, as RF is a comparative measure of equilibrium climate change and the Earth’s climate is not in an equilibrium state, additional climate change in the future is also expected from present-day RFs (see Sections 2.2 and 10.7 ). As previously stated in Section 2.2 ,RF alone is not a suitable metric for weighting emissions; for this purpose, the lifetime of the forcing agent also needs to be considered (see Sections 2.9.4 and 2.10 ).
RFs are considered external to the climate system (see Section 2.2 ). Aside from the natural RFs (solar, volcanoes), the other RFs are considered to be anthropogenic (i.e., directly attributable to human activities). For the LLGHGs it is assumed that all changes in their concentrations since pre-industrial times are human-induced (either directly through emissions or from land use changes); these concentration changes are used to calculate the RF. Likewise, stratospheric ozone changes are also taken from satellite observations and changes are primarily attributed to Montreal-Protocol controlled gases, although there may also be a climate feedback contribution to these trends (see Section 2.3.4 ). For the other RFs, anthropogenic emissions and/or human-induced land use changes are used in conjunction with CTMs and/or GCMs to estimate the anthropogenic RF.
2.9.1 Uncertainties in Radiative Forcing
The TAR assessed uncertainties in global mean RF by attaching an error bar to each RF term that was ‘guided by the range of published values and physical understanding’. It also quoted a level of scientific understanding (LOSU) for each RF, which was a subjective judgment of the estimate’s reliability.
The concept of LOSU has been slightly modified based on the IPCC Fourth Assessment Report (AR4) uncertainty guidelines. Error bars now represent the 5 to 95% (90%) confidence range (see Box TS.1). Only ‘well-established’ RFs are quantified. ‘Well established’ implies that there is qualitatively both sufficient evidence and sufficient consensus from published results to estimate a central RF estimate and a range. ‘Evidence’ is assessed by an A to C grade, with an A grade implying strong evidence and C insufficient evidence. Strong evidence implies that observations have verified aspects of the RF mechanism and that there is a sound physical model to explain the RF. ‘Consensus’ is assessed by assigning a number between 1 and 3, where 1 implies a good deal of consensus and 3 insufficient consensus. This ranks the number of studies, how well studies agree on quantifying the RF and especially how well observation-based studies agree with models. The product of ‘Evidence’ and ‘Consensus’ factors give the LOSU rank. These ranks are high, medium, medium-low, low or very low. Ranks of very low are not evaluated. The quoted 90% confidence range of RF quantifies the value uncertainty, as derived from the expert assessment of published values and their ranges. For most RFs, many studies have now been published, which generally makes the sampling of parameter space more complete and the value uncertainty more realistic, compared to the TAR. This is particularly true for both the direct and cloud albedo aerosol RF (see Section 2.4 ). Table 2.11 summarises the key certainties and uncertainties and indicates the basis for the 90% confidence range estimate. Note that the aerosol terms will have added uncertainties due to the uncertain semi-direct and cloud lifetime effects. These uncertainties in the response to the RF (efficacy) are discussed in Section 2.8.5 .
Table 2.11 indicates that there is now stronger evidence for most of the RFs discussed in this chapter. Some effects are not quantified, either because they do not have enough evidence or because their quantification lacks consensus. These include certain mechanisms associated with land use, stratospheric water vapour and cosmic rays. Cloud lifetime and the semi-direct effects are also excluded from this analysis as they are deemed to be part of the climate response (see Section 7.5 ). The RFs from the LLGHGs have both a high degree of consensus and a very large amount of evidence and, thereby, place understanding of these effects at a considerably higher level than any other effect.
Table 2.11 .Uncertainty assessment of forcing agents discussed in this chapter. Evidence for the forcing is given a grade (A to C), with A implying strong evidence and C insufficient evidence. The degree of consensus among forcing estimates is given a 1, 2 or 3 grade, where grade 1 implies a good deal of consensus and grade 3 implies an insufficient consensus. From these two factors, a level of scientific understanding is determined (LOSU). Uncertainties are in approximate order of importance with first-order uncertainties listed first.
| Evidence | Consensus | LOSU | Certainties | Uncertainties | Basis of RF range | |
|---|---|---|---|---|---|---|
| LLGHGs | A | 1 | High | Past and present concentrations; spectroscopy | Pre-industrial concentrations of some species; vertical profile in stratosphere; spectroscopic strength of minor gases | Uncertainty assessment of measured trends from different observed data sets and differences between radiative transfer models |
| Stratospheric ozone | A | 2 | Medium | Measured trends and its vertical profile since 1980; cooling of stratosphere; spectroscopy | Changes prior to 1970; trends near tropopause; effect of recent trends | Range of model results weighted to calculations employing trustworthy observed ozone trend data |
| Tropospheric ozone | A | 2 | Medium | Present-day concentration at surface and some knowledge of vertical and spatial structure of concentrations and emissions; spectroscopy | Pre-industrial values and role of changes in lightning; vertical structure of trends near tropopause; aspects of emissions and chemistry | Range of published model results, upper bound increased to account for anthropogenic trend in lightning |
| Stratospheric water vapour from CH4 | A | 3 | Low | Global trends since 1990; CH4 contribution to trend; spectroscopy | Global trends prior to 1990; radiative transfer in climate models; CTM models of CH4 oxidation | Range based on uncertainties in CH4 contribution to trend and published RF estimates |
| Direct aerosol | A | 2to 3 | Medium to Low | Ground-based and satellite observations; some source regions and modelling | Emission sources and their history vertical structure of aerosol, optical properties, mixing and separation from natural background aerosol | Range of published model results with allowances made for comparisons with satellite data |
| Cloud albedo effect (all aerosols) | B | 3 | Low | Observed in case studies – e.g., ship tracks; GCMs model an effect | Lack of direct observational evidence of a global forcing | Range of published model results and published results where models have been constrained by satellite data |
| Surface albedo (land use) | A | 2to 3 | Medium to Low | Some quantification of deforestation and desertification | Separation of anthropogenic changes from natural | Based on range of published estimates and published uncertainty analyses |
| Surface albedo (BC aerosol on snow) | B | 3 | Low | Estimates of BC aerosol on snow; some model studies suggest link | Separation of anthropogenic changes from natural; mixing of snow and BC aerosol; quantification of RF | Estimates based on a few published model studies |
| Persistent linear Contrails | A | 3 | Low | Cirrus radiative and microphysical properties; aviation emissions; contrail coverage in certain regions | Global contrail coverage and optical properties | Best estimate based on recent work and range from published model results |
Table 2.11 (continued)
| Evidence | Consensus | LOSU | Certainties | Uncertainties | Basis of RF range | |
|---|---|---|---|---|---|---|
| Solar irradiance | B | 3 | Low | Measurements over last 25 years; proxy indicators of solar activity | Relationship between proxy data and total solar irradiance; indirect ozone effects | Range from available reconstructions of solar irradiance and their qualitative assessment |
| Volcanic aerosol | A | 3 | Low | Observed aerosol changes from Mt. Pinatubo and El Chichón; proxy data for past eruptions; radiative effect of volcanic aerosol | Stratospheric aerosol concentrations from pre-1980 eruptions; atmospheric feedbacks | Past reconstructions/estimates of explosive volcanoes and observations of Mt. Pinatubo aerosol |
| Stratospheric water vapour from causes other than CH4 oxidation | C | 3 | Very Low | Empirical and simple model studies suggest link; spectroscopy | Other causes of water vapour trends poorly understood | Not given |
| Tropospheric water vapour from irrigation | C | 3 | Very Low | Process understood; spectroscopy; some regional information | Global injection poorly quantified | Not given |
| Aviation-induced cirrus | C | 3 | Very Low | Cirrus radiative and microphysical properties; aviation emissions; contrail coverage in certain regions | Transformation of contrails to cirrus; aviation’s effect on cirrus clouds | Not given |
| Cosmic rays | C | 3 | Very Low | Some empirical evidence and some observations as well as microphysical models suggest link to clouds | General lack/doubt regarding physical mechanism; dependence on correlation studies | Not given |
| Other surface effects | C | 3 | Very Low | Some model studies suggest link and some evidence of relevant processes | Quantification of RF and interpretation of results in forcing feedback context difficult | Not given |
2.9.2 Global Mean Radiative Forcing
The RFs discussed in this chapter, their uncertainty ranges and their efficacies are summarised in Figure 2.20 and Table 2.12. Radiative forcings from forcing agents have been combined into their main groupings. This is particularly useful for aerosol as its total direct RF is considerably better constrained than the RF from individual aerosol types (see Section 2.4.4 ). Table 2.1 gives a further component breakdown of RF for the LLGHGs. Radiative forcings are the stratospherically adjusted RF and they have not been multiplied by efficacies (see Sections 2.2 and 2.8 ).
In the TAR, no estimate of the total combined RF from all anthropogenic forcing agents was given because: a) some of the forcing agents did not have central or best estimates; b) a degree of subjectivity was included in the error estimates; and c) uncertainties associated with the linear additivity assumption and efficacy had not been evaluated. Some of these limitations still apply. However, methods for objectively adding the RF of individual species have been developed (e.g., Schwartz and Andreae, 1996 [JoC, ARC] ; Boucher and Haywood, 2001 [JoC, MoS, SRC] ). In addition, as efficacies are now better understood and quantified (see Section 2.8.5 ), and as the linear additivity assumption has been more thoroughly tested (see Section 2.8.4 ), it becomes scientifically justifiable for RFs from different mechanisms to be combined, with certain exceptions as noted below. Adding together the anthropogenic RF values shown in panel (A) of Figure 2.20 and combining their individual uncertainties gives the probability density functions (PDFs) of RF that are shown in panel (B). Three PDFs are shown: the combined RF from greenhouse gas changes (LLGHGs and ozone); the combined direct aerosol and cloud albedo RFs and the combination of all anthropogenic RFs. The solar RF is not included in any of these distributions. The PDFs are generated by combining the 90% confidence estimates for the RFs, assuming independence and employing a one-million point Monte Carlo simulation to derive the PDFs (see Boucher and Haywood, 2001 [JoC, MoS, SRC] ; and Figure 2.20 caption for details).
The PDFs show that LLGHGs and ozone contribute a positive RF of +2.9 ± 0.3 Wm–2.The combined aerosol direct and cloud albedo effect exert an RF that is virtually certain to be negative, with a median RF of –1.3 Wm–2 and a –2.2 to –0.5 Wm–2 90% confidence range. The asymmetry in the combined aerosol PDF is caused by the estimates in Tables 2.6 and 2.7 being non-Gaussian. The combined net RF estimate for all anthropogenic drivers has a value of +1.6 Wm–2 with a 0.6 to 2.4 Wm–2 90% confidence range. Note that the RFs from surface albedo change, stratospheric water vapour change and persistent contrails are only included in the combined anthropogenic PDF and not the other two.
Statistically, the PDF shown in Figure 2.20 indicates just a 0.2% probability that the total RF from anthropogenic agents is negative, which would suggest that it is virtually certain that the combined RF from anthropogenic agents is positive. Additionally, the PDF presented here suggests that it is extremely likely that the total anthropogenic RF is larger than +0.6 Wm–2.This combined anthropogenic PDF is better constrained than that shown in Boucher and Haywood 2001 [JoC, MoS, SRC] ) because each of the individual RFs have been quantified to 90% confidence levels, enabling a more definite assessment, and because the uncertainty in some of the RF estimates is considerably reduced. For example, modelling of the total direct RF due to aerosols is better constrained by satellite and surface-based observations( Section 2.4.2 ), and the current estimate of the cloud albedo indirect effect has a best estimate and uncertainty associated with it, rather than just a range. The LLGHG RF has also increased by 0.20 Wm–2 since 1998, making a positive RF more likely than in Boucher and Haywood 2001 [JoC, MoS, SRC] ).
Nevertheless, there are some structural uncertainties associated with the assumptions used in the construction of the PDF and the assumptions describing the component uncertainties. Normal distributions are assumed for most RF mechanisms (with the exceptions noted in the caption); this may not accurately capture extremes. Additionally, as in Boucher and Haywood 2001 [JoC, MoS, SRC] ), all of the individual RF mechanisms are given equal weighting, even though the level of scientific understanding differs between forcing mechanisms. Note also that variation in efficacy and hence the semi-direct and cloud lifetime effects are not accounted for, as these are not considered to be RFs in this report (see Section 2.2 ). Adding these effects, together with other potential mechanisms that have so far not been defined as RFs and quantified, would introduce further uncertainties but give a fuller picture of the role of anthropogenic drivers. Introducing efficacy would give a broader PDF and a large cloud lifetime effect would reduce the median estimate. Despite these caveats, from the current knowledge of individual forcing mechanisms presented here it remains extremely likely that the combined anthropogenic RF is both positive and substantial (best estimate: +1.6 Wm–2).
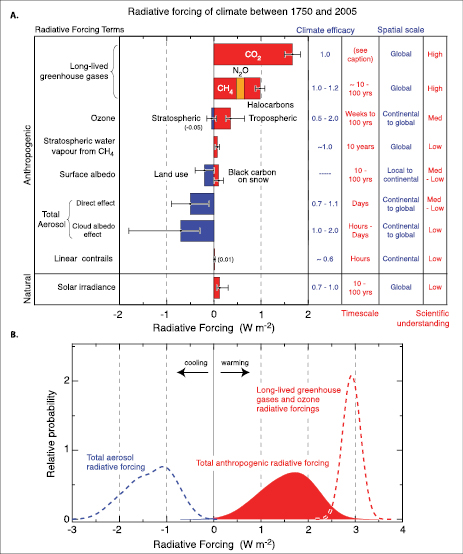
Figure 2.20. (A) Global mean RFs from the agents and mechanisms discussed in this chapter, grouped by agent type. Anthropogenic RFs and the natural direct solar RF are shown. The plotted RF values correspond to the bold values in Table 2.12. Columns indicate other characteristics of the RF; efficacies are not used to modify the RFs shown. Time scales represent the length of time that a given RF term would persist in the atmosphere after the associated emissions and changes ceased. No CO2 time scale is given, as its removal from the atmosphere involves a range of processes that can span long time scales, and thus cannot be expressed accurately with a narrow range of lifetime values. The scientific understanding shown for each term is described in Table 2.11 .(B) Probability distribution functions (PDFs) from combining anthropogenic RFs in (A). Three cases are shown: the total of all anthropogenic RF terms (block filled red curve; see also Table 2.12); LLGHGs and ozone RFs only (dashed red curve); and aerosol direct and cloud albedo RFs only (dashed blue curve). Surface albedo, contrails and stratospheric water vapour RFs are included in the total curve but not in the others. For all of the contributing forcing agents, the uncertainty is assumed to be represented by a normal distribution (and 90% confidence intervals) with the following exceptions: contrails, for which a lognormal distribution is assumed to account for the fact that the uncertainty is quoted as a factor of three; and tropospheric ozone, the direct aerosol RF (sulphate, fossil fuel organic and black carbon, biomass burning aerosols) and the cloud albedo RF, for which discrete values based on Figure 2.9, Table 2.6 and Table 2.7 are randomly sampled. Additional normal distributions are included in the direct aerosol effect for nitrate and mineral dust, as these are not explicitly accounted for in Table 2.6 .A one-million point Monte Carlo simulation was performed to derive the PDFs ( Boucher and Haywood, 2001 [JoC, MoS, SRC] ). Natural RFs (solar and volcanic) are not included in these three PDFs. Climate efficacies are not accounted for in forming the PDFs.
2.9.3 Global Mean Radiative Forcing by Emission Precursor
The RF due to changes in the concentration of a single forcing agent can have contributions from emissions of several compounds ( Shindell et al., 2005 [PoC, JoC, MoS, ARC] ). The RF of CH4,for example, is affected by CH4 emissions, as well as NOx emissions. The CH4 RF quoted in Table 2.12 and shown in Figure 2.20 is a value that combines the effects of both emissions. As an anthropogenic or natural emission can affect several forcing agents, it is useful to assess the current RF caused by each primary emission. For example, emission of NOx affects CH4,tropospheric ozone and tropospheric aerosols. Based on a development carried forward from the TAR, this section assesses the RF terms associated with each principal emission including indirect RFs related to perturbations of other forcing agents, with the results shown in Figure 2.21 .The following indirect forcing mechanisms are considered:
- fossil carbon from non-CO2 gaseous compounds, which eventually increase CO2 in the atmosphere (from CO, CH4,and NMVOC emissions);
- changes in stratospheric ozone (fromN2Oand halocarbon (CFCs, HCFC, halons, etc.) emissions);
- changes in tropospheric ozone (from CH4,NOx,CO, and NMVOC emissions);
- changes in OH affecting the lifetime of CH4 (from CH4,CO, NOx,and NMVOC emissions); and
- changing nitrate and sulphate aerosols through changes in NOx and SO2 emissions, respectively.
For some of the principal RFs (e.g., BC, land use and mineral dust) there is not enough quantitative information available to assess their indirect effects, thus their RFs are the same as those presented in Table 2.12. Table 2.5 gives the total (fossil and biomass burning) direct RFs for BC and organic carbon aerosols that are used to obtain the average shown in Figure 2.21 . Table 2.13 summarises the direct and indirect RFs presented in Figure 2.21 ,including the methods used for estimating the RFs and the associated uncertainty. Note that for indirect effects through changes in chemically active gases (e.g., OH or ozone), the emission-based RF is not uniquely defined since the effect of one precursor will be affected by the levels of the other precursors. The RFs of indirect effects on CH4 and ozone by NOx,CO and VOC emissions are estimated by removing the anthropogenic emissions of one precursor at a time. A sensitivity analysis by Shindell et al. 2005 [PoC, JoC, MoS, ARC] ) indicates that the nonlinear effect induced by treating the precursors separately is of the order of 10% or less. Very uncertain indirect effects are not included in Table 2.13 and Figure 2.21 .These include ozone changes due to solar effects, changes in secondary organic aerosols through changes in the ozone/OH ratio and apportioning of the cloud albedo changes to each aerosol type ( Hansen et al., 2005 [JoC, MoS, ARC] ).
Table 2.12. Global mean radiative forcings since 1750 and comparison with earlier assessments. Bold rows appear on Figure 2.20. The first row shows the combined anthropogenic RF from the probability density function in panel B of Figure 2.20. The sum of the individual RFs and their estimated errors are not quite the same as the numbers presented in this row due to the statistical construction of the probability density function.
| Global mean radiative forcing (Wm–2)a | ||||
|---|---|---|---|---|
| SAR (1750–1993) | TAR (1750–2005) | AR4 (1750-2005) | Summary comments on changes since the TAR | |
| Combined Anthropogenic RF | Not evaluated | Not evaluated | 1.6 [–1.0, +0.8] | Newly evaluated. Probabilitydensity function estimate |
| Long-lived Greenhouse gases (Comprising CO2,CH4,N2O, and halocarbons) | +2.45 [15%] (CO2 1.56; CH4 0.47;N2O0.14; Halocarbons 0.28) | +2.43 [10%] (CO2 1.46; CH4 0.48;N2O0.15; Halocarbons 0.34b) | +2.63 [±0.26] (CO2 1.66 [±0.17]; CH4 0.48 [±0.05];N2O0.16 [±0.02]; Halocarbons 0.34[±0.03]) | Total increase in RF, due toupward trends, particularly inCO2.Halocarbon RF trendis positiveb |
| Stratospheric ozone | –0.1 [2x] | –0.15 [67%] | –0.05 [±0.10] | Re-evaluated to be weaker |
| Tropospheric ozone | +0.40 [50%] | +0.35 [43%] | +0.35 [–0.1, +0.3] | Best estimate unchanged.However, a larger RF couldbe possible |
| Stratospheric water vapourfrom CH4 | Not evaluated | +0.01 to +0.03 | +0.07 [±0.05] | Re-evaluated to be higher |
| Total direct aerosol | Not evaluated | Not evaluated | –0.50 [±0.40] | Newly evaluated |
| Direct sulphate aerosol | –0.40 [2x] | –0.40 [2x] | –0.40 [±0.20] | Better constrained |
| Direct fossil fuel aerosol (organic carbon) | Not evaluated | –0.10 [3x] | –0.05 [±0.05] | Re-evaluated to be weaker |
| Direct fossil fuel aerosol (BC) | +0.10 [3x] | +0.20 [2x] | +0.20 [±0.15] | Similar best estimate to the TAR.Response affected by semi-direct effects |
| Direct biomass burning aerosol | –0.20 [3x] | –0.20 [3x] | +0.03 [±0.12] | Re-evaluated and sign changed.Response affected by semi-direct effects |
| Direct nitrate aerosol | Not evaluated | Not evaluated | –0.10 [±0.10] | Newly evaluated |
| Direct mineral dust aerosol | Not evaluated | –0.60 to +0.40 | –0.10 [±0.20] | Re-evaluated to have a smalleranthropogenic fraction |
| Cloud albedo effect | 0to –1.5 (sulphate only) | 0.0 to –2.0 (all aerosols) | –0.70 [–1.1, +0.4] (all aerosols) | Best estimate now given |
| Surface albedo (land use) | Not evaluated | –0.20 [100%] | –0.20 [±0.20] | Additional studies |
| Surface albedo (BC aerosol on snow) | Not evaluated | Not evaluated | +0.10 [±0.10] | Newly evaluated |
| Persistent linear contrails | Not evaluated | 0.02 [3.5x] | 0.01 [–0.007, +0.02] | Re-evaluated to be smaller |
| Solar irradiance | +0.30 [67%] | +0.30 [67%] | +0.12 [–0.06, +0.18] | Re-evaluated to be less than half |
Table. 2.13. Emission-based RFs for emitted components with radiative effects other than through changes in their atmospheric abundance. Minor effects where the estimated RF is less than 0.01 Wm–2 are not included. Effects on sulphate aerosols are not included since SO2 emission is the only significant factor affecting sulphate aerosols. Method of calculation and uncertainty ranges are given in the footnotes. Values represent RF in 2005 due to emissions and changes since 1750 . See Figure 2.21 for graphical presentation of these values.
| CO2 | CH4 | CFC/ HCFC | N2O | HFC/ PFC/SF6 | BC- direct | BC snow albedo | Organic carbon | O3(T)a | O3(S)b | H2O(S)c | Nitrate aerosols | Indirect cloud albedo effect | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Component emitted | Atmospheric or surface change directly causing radiative forcing | ||||||||||||
| CO2 | 1.56d | ||||||||||||
| CH4 | 0.016d | 0.57e | 0.2e | 0.07f | |||||||||
| CFC/HCFC/halons | 0.32g | –0.04h | |||||||||||
| N2O | 0.15g | –0.01h | |||||||||||
| HFC/PFC/SF6 | 0.017g | ||||||||||||
| CO/VOC | 0.06d | 0.08e | 0.13e | ||||||||||
| NOx | –0.17e | 0.06e | –0.10i | Xj | |||||||||
| BC | 0.34k | 0.1l | Xj | ||||||||||
| OC | –0.19k | Xj | |||||||||||
| SO2 | Xj | ||||||||||||
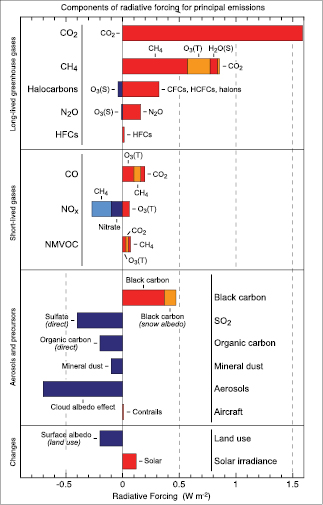
Figure 2.21. Components of RF for emissions of principal gases, aerosols and aerosol precursors and other changes. Values represent RF in 2005 due to emissions and changes since 1750 . (S) and (T) next to gas species represent stratospheric and tropospheric changes, respectively. The uncertainties are given in the footnotes to Table 2.13 .Quantitative values are displayed in Table 2.13 .
2.9.4 Future Climate Impact of Current Emissions
The changes in concentrations since pre-industrial time of the long-lived components causing the RF shown in Figure 2.20 are strongly influenced by the past history of emissions. A different perspective is obtained by integrating RF over a future time horizon for a one-year ‘pulse’ of global emissions (e.g., Jacobson 2002 [JoC] ) used this approach to compare fossil fuel organic and BC aerosols to CO2). Comparing the contribution from each forcing agent as shown in Figure 2.22 gives an indication of the future climate impact for current (year 2000 ) emissions of the different forcing agents. For the aerosols, the integrated RF is obtained based on the lifetimes, burdens and RFs from the AeroCom experiments, as summarised in Tables 2.4 and 2.5 .For ozone precursors (CO, NOx and NMVOCs), data are taken from Derwent et al. 2001 [JoC, ARC] Collins et al. 2002 [JoC, ARC] Stevenson et al. 2004 [JoC, MoS, ARC] and Berntsen et al. 2005 [JoC, SRC] ), while for the long-lived species the radiative efficiencies and lifetimes are used, as well as a response function for CO2 (see Section 2.10.2 , Table 2.14 ). Uncertainties in the estimates of the integrated RF originate from uncertainties in lifetimes, optical properties and current global emissions.
Figure 2.22 shows the integrated RF for both a 20- and 100-year time horizon. Choosing the longer time horizon of 100 years, as was done in the GWPs for the long-lived species included in the Kyoto Protocol, reduces the apparent importance of the shorter-lived species. It should be noted that the compounds with long lifetimes and short emission histories will tend to contribute more to the total with this ‘forward looking’ perspective than in the standard ‘IPCC RF bar chart diagram’( Figure 2.20 ).
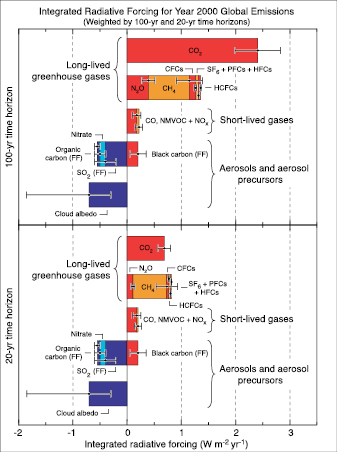
Figure 2.22. Integrated RF of year 2000 emissions over two time horizons (20 and 100 years). The figure gives an indication of the future climate impact of current emissions. The values for aerosols and aerosol precursors are essentially equal for the two time horizons. It should be noted that the RFs of short-lived gases and aerosol depend critically on both when and where they are emitted; the values given in the figure apply only to total global annual emissions. For organic carbon and BC, both fossil fuel (FF) and biomass burning emissions are included. The uncertainty estimates are based on the uncertainties in emission sources, lifetime and radiative efficiency estimates.
2.9.5 Time Evolution of Radiative Forcing and Surface Forcing
There is a good understanding of the time evolution of the LLGHG concentrations from in situ measurements over the last few decades and extending further back using firn and ice core data (see Section 2.3 , FAQ 2.1 ,Figure 1 and Chapter6 ). Increases in RF are clearly dominated by CO2.Halocarbon RF has grown rapidly since 1950, but the RF growth has been cut dramatically by the Montreal Protocol (see Section 2.3.4 ). The RF of CFCs is declining; in addition, the combined RF of all ozone-depleting substances (ODS) appears to have peaked at 0.32 Wm–2 during 2003 . However, substitutes for ODS are growing at a slightly faster rate, so halocarbon RF growth is still positive( Table 2.1 ). Although the trend in halocarbon RF since the time of the TAR has been positive (see Table 2.1 ), the halocarbon RF in this report, as shown in Table 2.12, is the same as in the TAR; this is due to a re-evaluation of the TAR results.
Radiative forcing time series for the natural (solar, volcanic aerosol) forcings are reasonably well known for the past 25 years; estimates further back are prone to uncertainties( Section 2.7 ). Determining the time series for aerosol and ozone RF is far more difficult because of uncertainties in the knowledge of past emissions and chemical-microphysical modelling. Several time series for these and other RFs have been constructed (e.g., Myhre et al., 2001 [MoS, SRC] ; Ramaswamy et al., 2001 [NPR, MoS, SRC] ; Hansen et al., 2002 [JoC, MoS, ARC] ). General Circulation Models develop their own time evolution of many forcings based on the temporal history of the relevant concentrations. As an example, the temporal evolution of the global and annual mean, instantaneous, all-sky RF and surface forcing due to the principal agents simulated by the Model for Interdisciplinary Research on Climate (MIROC) + Spectral Radiation-Transport Model for Aerosol Species (SPRINTARS) GCM ( Nozawa et al., 2005 [JoC, ARC] ; Takemura et al., 2005 [JoC, MoS, SRC] ) is illustrated in Figure 2.23 .Although there are differences between models with regards to the temporal reconstructions and thus present-day forcing estimates, they typically have a qualitatively similar temporal evolution since they often base the temporal histories on similar emissions data.
General Circulation Models compute the climate response based on the knowledge of the forcing agents and their temporal evolution. While most current GCMs incorporate the trace gas RFs, aerosol direct effects, solar and volcanoes, a few have in addition incorporated land use change and cloud albedo effect. While LLGHGs have increased rapidly over the past 20 years and contribute the most to the present RF (refer also to Figure 2.20 and FAQ 2.1 ,Figure 1), Figure 2.23 also indicates that the combined positive RF of the greenhouse gases exceeds the contributions due to all other anthropogenic agents throughout the latter half of the 20th century.
The solar RF has a small positive value. The positive solar irradiance RF is likely to be at least five times smaller than the combined RF due to all anthropogenic agents, and about an order of magnitude less than the total greenhouse gas contribution (Figures 2.20 and 2.23 and Table 2.12; see also the Foukal et al., 2006 [PoC, JoC, SRC] review). The combined natural RF consists of the solar RF plus the large but transitory negative RF from episodic, explosive volcanic eruptions of which there have been several over the past half century (see Figure 2.18 ). Over particularly the 1950 to 2005 period, the combined natural forcing has been either negative or slightly positive (less than approximately 0.2 Wm–2), reaffirming and extending the conclusions in the TAR. Therefore, it is exceptionally unlikely that natural RFs could have contributed a positive RF of comparable magnitude to the combined anthropogenic RF term over the period 1950 to 2005 ( Figure 2.23 ). Attribution studies with GCMs employ the available knowledge of the evolution of the forcing over the 20th century, and particularly the features distinguishing the anthropogenic from the natural agents (see also Section 9.2 ).
The surface forcing( Figure 2.23 ,top panel), in contrast to RF, is dominated by the strongly negative shortwave effect of the aerosols (tropospheric and the episodic volcanic ones), with the LLGHGs exerting a small positive effect. Quantitative values of the RFs and surface forcings by the agents differ across models in view of the differences in model physics and in the formulation of the forcings due to the short-lived species (see Section 10.2 ,Collins et al. 2006 [JoC, MoS, SRC] and Forster and Taylor 2006 [JoC, MoS, SRC] ) for further discussion on uncertainties in GCMs’ calculation of RF and surface forcing). As for RF, it is difficult to specify uncertainties in the temporal evolution, as emissions and concentrations for all but the LLGHGs are not well constrained.
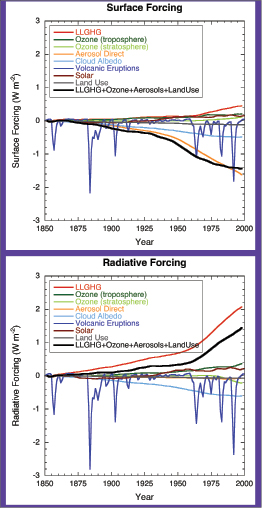
Figure 2.23. Globally and annually averaged temporal evolution of the instantaneous all-sky RF (top panel) and surface forcing (bottom panel) due to various agents, as simulated in the MIROC+SPRINTARS model ( Nozawa et al., 2005 [JoC, ARC] ; Takemura et al., 2005 [JoC, MoS, SRC] ). This is an illustrative example of the forcings as implemented and computed in one of the climate models participating in the AR4. Note that there could be differences in the RFs among models. Most models simulate roughly similar evolution of the LLGHGs’ RF.
2.9.6 Spatial Patterns of Radiative Forcing and Surface Forcing
Figure 6.7 of Ramaswamy et al. 2001 [NPR, MoS, SRC] ) presented examples of the spatial patterns for most of the RF agents discussed in this chapter; these examples still hold. Many of the features seen in Figure 6.7 of Ramaswamy et al. 2001 [NPR, MoS, SRC] ) are generic. However, additional uncertainties exist for the spatial patterns compared to those for the global-mean RF. Spatial patterns of the aerosol RF exhibit some of the largest differences between models, depending on the specification of the aerosols and their properties, and whether or not indirect cloud albedo effects are included. The aerosol direct and cloud albedo effect RF also depend critically on the location of clouds, which differs between the GCMs. Figure 2.24 presents illustrative examples of the spatial pattern of the instantaneous RF between 1860 and present day, due to natural plus anthropogenic agents, from two GCMs. Volcanic aerosols play a negligible role in this calculation owing to the end years considered and their virtual absence during these years. The MIROC+SPRINTARS model includes an aerosol cloud albedo effect while the Geophysical Fluid Dynamics Laboratory Coupled Climate Model (GFDL CM2.1) ( Delworth et al., 2005 [JoC, MoS, SRC] ; Knutson et al., 2006 [JoC, MoS, ARC] ) does not. Radiative forcing over most of the globe is positive and is dominated by the LLGHGs. This is more so for the SH than for the NH, owing to the pronounced aerosol presence in the mid-latitude NH (see also Figure 2.12 ), with the regions of substantial aerosol RF clearly manifest over the source-rich continental areas. There are quantitative differences between the two GCMs in the global mean RF, which are indicative of the uncertainties in the RF from the non-LLGHG agents, particularly aerosols (see Section 2.4 and Figure 2.12 d). The direct effect of aerosols is seen in the total RF of the GFDL model over NH land regions, whereas the cloud albedo effect dominates the MIROC+SPRINTARS model in the stratocumulus low-latitude ocean regions. Note that the spatial pattern of the forcing is not indicative of the climate response pattern.
Wherever aerosol presence is considerable (namely the NH), the surface forcing is negative, relative to pre-industrial times( Figure 2.24 ). Because of the aerosol influence on the reduction of the shortwave radiation reaching the surface (see also Figure 2.12 f), there is a net (sum of shortwave and longwave) negative surface forcing over a large part of the globe (see also Figure 2.23 ). In the absence of aerosols, LLGHGs increase the atmospheric longwave emission, with an accompanying increase in the longwave radiative flux reaching the surface. At high latitudes and in parts of the SH, there are fewer anthropogenic aerosols and thus the surface forcing has a positive value, owing to the LLGHGs.
These spatial patterns of RF and surface forcing imply different changes in the NH equator-to-pole gradients for the surface and tropopause. These, in turn, imply different changes in the amount of energy absorbed by the troposphere at low and high latitudes. The aerosol influences are also manifest in the difference between the NH and SH in both RF and surface forcing.
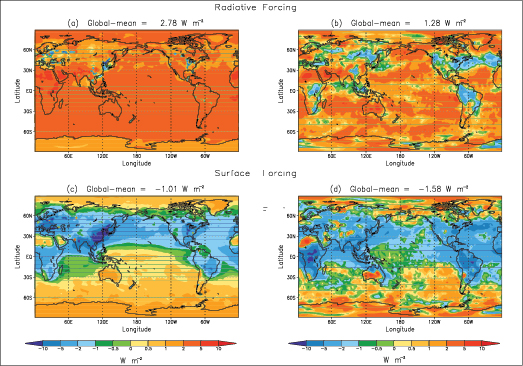
Figure 2.24. Instantaneous change in the spatial distribution of the net (solar plus longwave) radiative flux (W m–2)due to natural plus anthropogenic forcings between the years 1860 and 2000 . Results here are intended to be illustrative examples of these quantities in two different climate models. (a) and (c) correspond to tropopause and surface results using the GFDL CM 2.1 model (adapted from Knutson et al., 2006 [JoC, MoS, ARC] ). (b) and (d) correspond to tropopause and surface results using the MIROC+SPRINTARS model (adapted from Nozawa et al., 2005 [JoC, ARC] and Takemura et al., 2005 [JoC, MoS, SRC] ). Note that the MIROC+SPRINTARS model takes into account the aerosol cloud albedo effect while the CM 2.1 model does not.
2.10 Global Warming Potentials and Other Metrics for Comparing Different Emissions
2.10.1 Definition of an Emission Metric and the Global Warming Potential
Multi-component abatement strategies to limit anthropogenic climate change need a framework and numerical values for the trade-off between emissions of different forcing agents. Global Warming Potentials or other emission metrics provide a tool that can be used to implement comprehensive and cost-effective policies (Article 3 of the UNFCCC) in a decentralised manner so that multi-gas emitters (nations, industries) can compose mitigation measures, according to a specified emission constraint, by allowing for substitution between different climate agents. The metric formulation will differ depending on whether a long-term climate change constraint has been set (e.g., Manne and Richels, 2001 [JoC] ) or no specific long-term constraint has been agreed upon (as in the Kyoto Protocol). Either metric formulation requires knowledge of the contribution to climate change from emissions of various components over time. The metrics assessed in this report are purely physically based. However, it should be noted that many economists have argued that emission metrics need also to account for the economic dimensions of the problem they are intended to address (e.g., Bradford, 2001 [JoC] ; Manne and Richels, 2001 [JoC] ; Godal, 2003 [JoC] ; O’Neill, 2003 [JoC] ). Substitution of gases within an international climate policy with a long-term target that includes economic factors is discussed in Chapter3 of IPCC WGIII AR4. Metrics based on this approach will not be discussed in this report.
Avery general formulation of an emission metric is given by (e.g., ( Kandlikar, 1996 ) ):
AMi = ∫∞0 [(I(DC (r + i) (t)) – I(DCr (t))) x g(t)]dt
where I(∆Ci(t)) is a function describing the impact (damage and benefit) of change in climate (∆C) at time t.The expression g(t) is a weighting function over time (e.g., g(t)= e–kt is a simple discounting giving short-term impacts more weight) ( Heal, 1997 [JoC] ; Nordhaus, 1997 [JoC] ). The subscript r refers to a baseline emission path. For two emission perturbations i and j the absolute metric values AMi and AMj can be calculated to provide a quantitative comparison of the two emission scenarios. In the special case where the emission scenarios consist of only one component (as for the assumed pulse emissions in the definition of GWP), the ratio between AMi and AMj can be interpreted as a relative emission index for component i versus a reference component j (such as CO2 in the case of GWP).
There are several problematic issues related to defining a metric based on the general formulation given above ( Fuglestvedt et al., 2003 [JoC, MoS] ). A major problem is to define appropriate impact functions, although there have been some initial attempts to do this for a range of possible climate impacts ( Hammitt et al., 1996 [JoC] ; Tol, 2002 [MoS] ;( den Elzen et al., 2005 ) ). Given that impact functions can be defined, AM calculations would require regionally resolved climate change data (temperature, precipitation, winds, etc.) that would have to be based on GCM results with their inherent uncertainties ( Shine et al., 2005a [JoC, SRC] ). Other problematic issues include the definition of the temporal weighting function g(t) and the baseline emission scenarios.
Due to these difficulties, the simpler and purely physical GWP index, based on the time-integrated global mean RF of a pulse emission of 1 kg of some compound(i)relative to that of 1 kg of the reference gas CO2,was developed ( IPCC, 1990 [NPR] ) and adopted for use in the Kyoto Protocol. The GWP of component i is defined by

where TH is the time horizon, RFi is the global mean RF of component i, aiis the RF per unit mass increase in atmospheric abundance of component i (radiative efficiency),[Ci(t)] is the time-dependent abundance of i,and the corresponding quantities for the reference gas(r)in the denominator. The numerator and denominator are called the absolute global warming potential (AGWP) of i and r respectively. All GWPs given in this report use CO2 as the reference gas. The simplifications made to derive the standard GWP index include (1) setting g(t)= 1 (i.e., no discounting) up until the time horizon(TH)and then g(t) = 0 thereafter, (2) choosing a 1-kg pulse emission, (3) defining the impact function, I(∆C), to be the global mean RF, (4) assuming that the climate response is equal for all RF mechanisms and (5) evaluating the impact relative to a baseline equal to current concentrations (i.e., setting I(∆Cr(t)) = 0). The criticisms of the GWP metric have focused on all of these simplifications (e.g., O’Neill, 2000 [JoC] ; Smith and Wigley, 2000 [PoC, JoC, ARC] ; Bradford, 2001 [JoC] ; Godal, 2003 [JoC] ). However, as long as there is no consensus on which impact function(I(∆C))and temporal weighting functions to use (both involve value judgements), it is difficult to assess the implications of the simplifications objectively ( O’Neill, 2000 [JoC] ; Fuglestvedt et al., 2003 [JoC, MoS] ).
The adequacy of the GWP concept has been widely debated since its introduction ( O’Neill, 2000 [JoC] ; Fuglestvedt et al., 2003 [JoC, MoS] ). By its definition, two sets of emissions that are equal in terms of their total GWP-weighted emissions will not be equivalent in terms of the temporal evolution of climate response ( Fuglestvedt et al., 2000 [JoC, SRC] ; Smith and Wigley, 2000 [PoC, JoC, ARC] ). Using a 100-year time horizon as in the Kyoto Protocol, the effect of current emissions reductions (e.g., during the first commitment period under the Kyoto Protocol) that contain a significant fraction of short-lived species (e.g., CH4)will give less temperature reductions towards the end of the time horizon, compared to reductions in CO2 emissions only. Global Warming Potentials can really only be expected to produce identical changes in one measure of climate change – integrated temperature change following emissions impulses – and only under a particular set of assumptions ( O’Neill, 2000 [JoC] ). The Global Temperature Potential (GTP) metric (see Section 2.10.4.2 )provides an alternative approach by comparing global mean temperature change at the end of a given time horizon. Compared to the GWP, the GTP gives equivalent climate response at a chosen time, while putting much less emphasis on near-term climate fluctuations caused by emissions of short-lived species (e.g., CH4). However, as long as it has not been determined, neither scientifically, economically nor politically, what the proper time horizon for evaluating ‘dangerous anthropogenic interference in the climate system’ should be, the lack of temporal equivalence does not invalidate the GWP concept or provide guidance as to how to replace it. Although it has several known shortcomings, a multi-gas strategy using GWPs is very likely to have advantages over a CO2-only strategy ( O’Neill, 2003 [JoC] ). Thus, GWPs remain the recommended metric to compare future climate impacts of emissions of long-lived climate gases.
Globally averaged GWPs have been calculated for short-lived species, for example, ozone precursors and absorbing aerosols ( Fuglestvedt et al., 1999 [MoS] ; Derwent et al., 2001 [JoC, ARC] ; Collins et al., 2002 [JoC, ARC] ; Stevenson et al., 2004 [JoC, MoS, ARC] ; Berntsen et al., 2005 [JoC, SRC] ;( Bond and Sun, 2005 ) ). There might be substantial co-benefits realised in mitigation actions involving short-lived species affecting climate and air pollutants ( Hansen and Sato, 2004 [JoC, ARC] ); however, the effectiveness of the inclusion of short-lived forcing agents in international agreements is not clear ( (Rypdal et al., 2005 ) ). To assess the possible climate impacts of short-lived species and compare those with the impacts of the LLGHGs, a metric is needed. However, there are serious limitations to the use of global mean GWPs for this purpose. While the GWPs of the LLGHGs do not depend on location and time of emissions, the GWPs for short-lived species will be regionally and temporally dependent. The different response of precipitation to an aerosol RF compared to a LLGHG RF also suggests that the GWP concept may be too simplistic when applied to aerosols.
2.10.2 Direct Global Warming Potentials
All GWPs depend on the AGWP for CO2 (the denominator in the definition of the GWP). The AGWP of CO2 again depends on the radiative efficiency for a small perturbation of CO2 from the current level of about 380 ppm. The radiative efficiency per kilogram of CO2 has been calculated using the same expression as for the CO2 RF in Section 2.3.1 ,with an updated background CO2 mixing ratio of 378 ppm. For a small perturbation from 378 ppm, the RF is 0.0 1413 Wm–2 ppm–1 (8.7% lower than the TAR value). The CO2 response function (see Table 2.14 )is based on an updated version of the Bern carbon cycle model (Bern2.5CC; Joos et al. 2001 [PoC, JoC, SRC] ), using a background CO2 concentration of 378 ppm. The increased background concentration of CO2 means that the airborne fraction of emitted CO2 ( Section 7.3 )is enhanced, contributing to an increase in the AGWP for CO2.The AGWP values for CO2 for 20, 100, and 500 year time horizons are 2.47 × 10–14,8.69 × 10–14,and 28.6 × 10–14 Wm–2 yr (kg CO2)–1,respectively. The uncertainty in the AGWP for CO2 is estimated to be ±15%, with equal contributions from the CO2 response function and the RF calculation.
Updated radiative efficiencies for well-mixed greenhouse gases are given in Table 2.14 .Since the TAR, radiative efficiencies have been reviewed by Montzka et al. 2003 [NPR, SRC] and Velders et al. 2005 [NPR] Gohar et al. 2004 [JoC, MoS, SRC] and Forster et al. 2005 [MoS, SRC] ) investigated HFC compounds, with up to 40% differences from earlier published results. Based on a variety of radiative transfer codes, they found that uncertainties could be reduced to around 12% with well-constrained experiments. The HFCs studied were HFC-23, HFC-32, HFC-134a and HFC-227ea. Hurley et al. 2005 [JoC, MoS] ) studied the infrared spectrum and RF of perfluoromethane (CΩF4)and derived a 30% higher GWP value than given in the TAR. The RF calculations for the GWPs for CH4,N2Oand halogen-containing well-mixed greenhouse gases employ the simplified formulas given in Ramaswamy et al. 2001 [NPR, MoS, SRC] ; see Table 6.2 of the TAR). Table 2.14 gives GWP values for time horizons of 20, 100 and 500 years. The species in Table 2.14 are those for which either significant concentrations or large trends in concentrations have been observed or a clear potential for future emissions has been identified. The uncertainties of these direct GWPs are taken to be ±35% for the 5 to 95% (90%) confidence range.
Table 2.14 .Lifetimes, radiative efficiencies and direct (except for CH4)GWPs relative to CO2.For ozone-depleting substances and their replacements, data are taken from IPCC/TEAP ( 2005 ) unless otherwise indicated.
| Global Warming Potential for Given Time Horizon | |||||||
|---|---|---|---|---|---|---|---|
| Industrial Designation or Common Name (years) | Chemical Formula | Lifetime (years) | RadiativeEfficiency (Wm–2 ppb–1) | SAR‡ (100-yr) | 20-yr | 100-yr | 500-yr |
| Carbon dioxide | CO2 | See belowa | b1.4x10–5 | 1 | 1 | 1 | 1 |
| Methanec | CH4 | 12c | 3.7x10–4 | 21 | 72 | 25 | 7.6 |
| Nitrous oxide | N2O | 114 | 3.03x10–3 | 310 | 289 | 298 | 153 |
| Substances controlled by the Montreal Protocol | |||||||
| CFC-11 | CCl3F | 45 | 0.25 | 3,800 | 6,730 | 4,750 | 1,620 |
| CFC-12 | CCl2F2 | 100 | 0.32 | 8,100 | 11,000 | 10,900 | 5,200 |
| CFC-13 | CClF3 | 640 | 0.25 | 10,800 | 14,400 | 16,400 | |
| CFC-113 | CCl2FCClF2 | 85 | 0.3 | 4,800 | 6,540 | 6,130 | 2,700 |
| CFC-114 | CClF2CClF2 | 300 | 0.31 | 8,040 | 10,000 | 8,730 | |
| CFC-115 | CClF2CF3 | 1,700 | 0.18 | 5,310 | 7,370 | 9,990 | |
| Halon-1301 | CBrF3 | 65 | 0.32 | 5,400 | 8,480 | 7,140 | 2,760 |
| Halon-1211 | CBrClF2 | 16 | 0.3 | 4,750 | 1,890 | 575 | |
| Halon-2402 | CBrF2CBrF2 | 20 | 0.33 | 3,680 | 1,640 | 503 | |
| Carbon tetrachloride | CCl4 | 26 | 0.13 | 1,400 | 2,700 | 1,400 | 435 |
| Methyl bromide | CH3Br | 0.7 | 0.01 | 17 | 5 | 1 | |
| Methyl chloroform | CH3CCl3 | 5 | 0.06 | 506 | 146 | 45 | |
| HCFC-22 | CHClF2 | 12 | 0.2 | 1,500 | 5,160 | 1,810 | 549 |
| HCFC-123 | CHCl2CF3 | 1.3 | 0.14 | 90 | 273 | 77 | 24 |
| HCFC-124 | CHClFCF3 | 5.8 | 0.22 | 470 | 2,070 | 609 | 185 |
| HCFC-141b | CH3CCl2F | 9.3 | 0.14 | 2,250 | 725 | 220 | |
| HCFC-142b | CH3CClF2 | 17.9 | 0.2 | 1,800 | 5,490 | 2,310 | 705 |
| HCFC-225ca | CHCl2CF2CF3 | 1.9 | 0.2 | 429 | 122 | 37 | |
| HCFC-225cb | CHClFCF2CClF2 | 5.8 | 0.32 | 2,030 | 595 | 181 | |
| Hydrofluorocarbons | |||||||
| HFC-23 | CHF3 | 270 | 0.19 | 11,700 | 12,000 | 14,800 | 12,200 |
| HFC-32 | CH2F2 | 4.9 | 0.11 | 650 | 2,330 | 675 | 205 |
| HFC-125 | CHF2CF3 | 29 | 0.23 | 2,800 | 6,350 | 3,500 | 1,100 |
| HFC-134a | CH2FCF3 | 14 | 0.16 | 1,300 | 3,830 | 1,430 | 435 |
| HFC-143a | CH3CF3 | 52 | 0.13 | 3,800 | 5,890 | 4,470 | 1,590 |
| HFC-152a | CH3CHF2 | 1.4 | 0.09 | 140 | 437 | 124 | 38 |
| HFC-227ea | CF3CHFCF3 | 34.2 | 0.26 | 2,900 | 5,310 | 3,220 | 1,040 |
| HFC-236fa | CF3CH2CF3 | 240 | 0.28 | 6,300 | 8,100 | 9,810 | 7,660 |
| HFC-245fa | CHF2CH2CF3 | 7.6 | 0.28 | 3,380 | 1030 | 314 | |
| HFC-365mfc | CH3CF2CH2CF3 | 8.6 | 0.21 | 2,520 | 794 | 241 | |
| HFC-43-10mee | CF3CHFCHFCF2CF3 | 15.9 | 0.4 | 1,300 | 4,140 | 1,640 | 500 |
| Perfluorinated compounds | |||||||
| Sulphur hexafluoride | SF6 | 3,200 | 0.52 | 23,900 | 16,300 | 22,800 | 32,600 |
| Nitrogen trifluoride | NF3 | 740 | 0.21 | 12,300 | 17,200 | 20,700 | |
| PFC-14 | CF4 | 50,000 | 0.10 | 6,500 | 5,210 | 7,390 | 11,200 |
| PFC-116 | C2F6 | 10,000 | 0.26 | 9,200 | 8,630 | 12,200 | 18,200 |
Table 2.14 (continued)
| Global Warming Potential for Given Time Horizon | |||||||
|---|---|---|---|---|---|---|---|
| Industrial Designation or Common Name (years) | Chemical Formula | Lifetime (years) | RadiativeEfficiency (W m–2 ppb–1) | SAR‡ (100-yr) | 20-yr | 100-yr | 500-yr |
| Perfluorinated compounds (continued) | |||||||
| PFC-218 | 2,600 | 0.26 | 7,000 | 6,310 | 8,830 | 12,500 | |
| PFC-318 | 3,200 | 0.32 | 8,700 | 7,310 | 10,300 | 14,700 | |
| PFC-3-1-10 | 2,600 | 0.33 | 7,000 | 6,330 | 8,860 | 12,500 | |
| PFC-4-1-12 | 4,100 | 0.41 | 6,510 | 9,160 | 13,300 | ||
| PFC-5-1-14 | 3,200 | 0.49 | 7,400 | 6,600 | 9,300 | 13,300 | |
| PFC-9-1-18 | >1,000d | 0.56 | >5,500 | >7,500 | >9,500 | ||
| trifluoromethyl sulphur pentafluoride | 800 | 0.57 | 13,200 | 17,700 | 21,200 | ||
| Fluorinated ethers | |||||||
| HFE-125 | 136 | 0.44 | 13,800 | 14,900 | 8,490 | ||
| HFE-134 | 26 | 0.45 | 12,200 | 6,320 | 1,960 | ||
| HFE-143a | 4.3 | 0.27 | 2,630 | 756 | 230 | ||
| HCFE-235da2 | 2.6 | 0.38 | 1,230 | 350 | 106 | ||
| HFE-245cb2 | 5.1 | 0.32 | 2,440 | 708 | 215 | ||
| HFE-245fa2 | 4.9 | 0.31 | 2,280 | 659 | 200 | ||
| HFE-254cb2 | 2.6 | 0.28 | 1,260 | 359 | 109 | ||
| HFE-347mcc3 | 5.2 | 0.34 | 1,980 | 575 | 175 | ||
| HFE-347pcf2 | 7.1 | 0.25 | 1,900 | 580 | 175 | ||
| HFE-356pcc3 | 0.33 | 0.93 | 386 | 110 | 33 | ||
| HFE-449sl (HFE-7100) | 3.8 | 0.31 | 1,040 | 297 | 90 | ||
| HFE-569sf2 (HFE-7200) | 0.77 | 0.3 | 207 | 59 | 18 | ||
| HFE-43-10pccc124 (H-Galden 1040x) | 6.3 | 1.37 | 6,320 | 1,870 | 569 | ||
| HFE-236ca12 (HG-10) | 12.1 | 0.66 | 8,000 | 2,800 | 860 | ||
| HFE-338pcc13 (HG-01) | 6.2 | 0.87 | 5,100 | 1,500 | 460 | ||
| Perfluoropolyethers | |||||||
| PFPMIE | 800 | 0.65 | 7,620 | 10,300 | 12,400 | ||
| Hydrocarbons and other compounds – Direct Effects | |||||||
| Dimethylether | 0.015 | 0.02 | 1 | 1 | <<1 | ||
| Methylene chloride | 0.38 | 0.03 | 31 | 8.7 | 2.7 | ||
| Methyl chloride | 1.0 | 0.01 | 45 | 13 | 4 | ||
![]()
g Young et al. 2006 [NotFound] )
2.10.3 Indirect GWPs
Indirect radiative effects include the direct effects of degradation products or the radiative effects of changes in concentrations of greenhouse gases caused by the presence of the emitted gas or its degradation products. Direct effects of degradation products for the greenhouse gases are not considered to be significant ( WMO, 2003 [NPR] ). The indirect effects discussed here are linked to ozone formation or destruction, enhancement of stratospheric water vapour, changes in concentrations of the OH radical with the main effect of changing the lifetime of CH4,and secondary aerosol formation. Uncertainties for the indirect GWPs are generally much higher than for the direct GWPs. The indirect GWP will in many cases depend on the location and time of the emissions. For some species (e.g., NOx)the indirect effects can be of opposite sign, further increasing the uncertainty of the net GWP. This can be because background levels of reactive species (e.g., NOx)can affect the chemical response nonlinearly, and/or because the lifetime or the radiative effects of short-lived secondary species formed can be regionally dependent. Thus, the usefulness of the global mean GWPs to inform policy decisions can be limited. However, they are readily calculable and give an indication of the total potential of mitigating climate change by including a certain forcing agent in climate policy. Following the approach taken by the SAR and the TAR, the CO2 produced from oxidation of CH4,CO and NMVOCs of fossil origin is not included in the GWP estimates since this carbon has been included in the national CO2 inventories. This issue may need to be reconsidered as inventory guidelines are revised.
2.10.3.1 Methane
Four indirect radiative effects of CH4 emissions have been identified (see Prather et al., 2001 [NPR, ARC] ; Ramaswamy et al., 2001 [NPR, MoS, SRC] ). Methane enhances its own lifetime through changes in the OH concentration: it leads to changes in tropospheric ozone, enhances stratospheric water vapour levels, and produces CO2.The GWP given in Table 2.14 includes the first three of these effects. The lifetime effect is included by adopting a perturbation lifetime of 12 years (see Section 7.4 ). The effect of ozone production is still uncertain, and as in the TAR, it is included by enhancing the net of the direct and the lifetime effect by 25%. The estimate of RF caused by an increase in stratospheric water vapour has been increased significantly since the TAR (see Section 2.3.7 ). This has also been taken into account in the GWP estimate for CH4 by increasing the enhancement factor from 5% (TAR) to 15%. As a result, the 100-year GWP for CH4 has increased from 23 in the TAR to 25.
2.10.3.2 Carbon Monoxide
The indirect effects of CO occur through reduced OH levels (leading to enhanced concentrations of CH4)and enhancement of ozone. The TAR gave a range of 1.0 to 3.0 for the 100-year GWP. Since the TAR, Collins et al. 2002 [JoC, ARC] and Berntsen et al. 2005 [JoC, SRC] ) have calculated GWPs for CO emissions that range between 1.6 and 2.0, depending on the location of the emissions. Berntsen et al. 2005 [JoC, SRC] ) found that emissions of CO from Asia had a 25% higher GWP compared to European emissions. Averaging over the TAR values and the new estimates give a mean of 1.9 for the 100-year GWP for CO.
Table 2.15. Indirect GWPs (100-year) for 10 NMVOCs from Collins et al. 2002 [JoC, ARC] ) and for NOx emissions (on N-basis) from Derwent et al. 2001 [JoC, ARC] Wild et al. 2001 [JoC, ARC] Berntsen et al. 2005 [JoC, SRC] and Stevenson et al. 2004 [JoC, MoS, ARC] ). The second and third columns respectively represent the methane and ozone contribution to the net GWP and the fourth column represents the net GWP.
| Organic Compound/Study | GWPCH4 | GWPO3 | GWP |
|---|---|---|---|
| Ethane (C2H6) | 2.9 | 2.6 | 5.5 |
| Propane (C3H8) | 2.7 | 0.6 | 3.3 |
| Butane (C4H10) | 2.3 | 1.7 | 4.0 |
| Ethylene (C2H4) | 1.5 | 2.2 | 3.7 |
| Propylene (C3H6) | –2.0 | 3.8 | 1.8 |
| Toluene (C7H8) | 0.2 | 2.5 | 2.7 |
| Isoprene (C5H8) | 1.1 | 1.6 | 2.7 |
| Methanol (CH3OH) | 1.6 | 1.2 | 2.8 |
| Acetaldehyde (CH3CHO) | –0.4 | 1.7 | 1.3 |
| Acetone (CH3COCH3) | 0.3 | 0.2 | 0.5 |
| Derwent et al. NH surface NOxa,b | –24 | 11 | –12 |
| Derwent et al. SH surface NOxa,b | –64 | 33 | –31 |
| Wild et al., industrial NOx | –44 | 32 | –12 |
| Berntsen et al., surface NOx Asia | –31 to –42c | 55 to 70c | 25 to 29c |
| Berntsen et al., surface NOx Europe | –8.6 to –11c | 8.1 to 12.7 | –2.7 to +4.1c |
| Derwent et al., Aircraft NOxa,b | –145 | 246 | 100 |
| Wild et al., Aircraft NOx | –210 | 340 | 130 |
| Stevenson et al. Aircraft NOx | –159 | 155 | –3 |
2.10.3.3 Non-methane Volatile Organic Compounds
Collins et al. 2002 [JoC, ARC] ) calculated indirect GWPs for 10 NMVOCs with a global three-dimensional Lagrangian chemistry-transport model. Impacts on tropospheric ozone, CH4 (through changes in OH) and CO2 have been considered, using either an ‘anthropogenic’ emission distribution or a ‘natural’ emission distribution depending on the main sources for each gas. The indirect GWP values are given in Table 2.15. Weighting these GWPs by the emissions of the respective compounds gives a weighted average 100-year GWP of 3.4. Due to their short lifetimes and the nonlinear chemistry involved in ozone and OH chemistry, there are significant uncertainties in the calculated GWP values. Collins et al. 2002 [JoC, ARC] ) estimated an uncertainty range of –50% to +100%.
2.10.3.4 Nitrogen Oxides
The short lifetime and complex nonlinear chemistry, which cause two opposing indirect effects through ozone enhancements and CH4 reductions, make calculations of GWP for NOx emissions very uncertain ( Shine et al., 2005a [JoC, SRC] ). In addition, the effect of nitrate aerosol formation (see Section 2.4.4.5 ), which has not yet been included in model studies calculating GWPs for NOx,can be significant. Due to the nonlinear chemistry, the net RF of NOx emissions will depend strongly on the location of emission and, with a strict definition of a pulse emission for the GWP, also on timing (daily, seasonal) of the emissions ( Fuglestvedt et al., 1999 [MoS] ; Derwent et al., 2001 [JoC, ARC] ; Wild et al., 2001 [JoC, ARC] ; Stevenson et al., 2004 [JoC, MoS, ARC] ; Berntsen et al., 2005 [JoC, SRC] , 2006 ). Due to the lack of agreement even on the sign of the global mean GWP for NOx among the different studies and the omission of the nitrate aerosol effect, a central estimate for the 100-year GWP for NOx is not presented.
Chlorine- and bromine-containing halocarbons lead to ozone depletion when the halocarbon molecules are broken down in the stratosphere and chlorine or bromine atoms are released. Indirect GWPs for ozone-depleting halocarbons are estimated in Velders et al. 2005 [NPR] ; their Table 2.7 ). These are based on observed ozone depletion between 1980 and 1990 for 2005 emissions using the Daniel et al. 1995 [PoC, JoC, MoS, ARC] ) formulation. Velders et al. 2005 [NPR] ) did not quote net GWPs, pointing out that the physical characteristics of the CFC warming effect and ozone cooling effect were very different from each other.
2.10.3.6 Hydrogen
The main loss of hydrogen (H2)is believed to be through surface deposition, but about 25% is lost through oxidation by OH. In the stratosphere, this enhances the water vapour concentrations and thus also affects the ozone concentrations. In the troposphere, the chemical effects are similar to those of CO, leading to ozone production and CH4 enhancements ( Prather, 2003 [JoC, ARC] Derwent et al. 2001 [JoC, ARC] ) calculated an indirect 100-year GWP for the tropospheric effects ofH2 of 5.8, which includes the effects of CH4 lifetime and tropospheric ozone.
While the GWP is a simple and straightforward index to apply for policy makers to rank emissions of different greenhouse gases, it is not obvious on what basis ‘equivalence’ between emissions of different species is obtained ( Smith and Wigley, 2000 [PoC, JoC, ARC] ; Fuglestvedt et al., 2003 [JoC, MoS] ). The GWP metric is also problematic for short-lived gases or aerosols (e.g., NOx or BC aerosols), as discussed above. One alternative, the RF index (RFI) introduced by IPCC 1999 [NPR] ), should not be used as an emission metric since it does not account for the different residence times of different forcing agents.
2.10.4 New Alternative Metrics for Assessing Emissions
2.10.4.1 Revised GWP Formulations
As discussed in Section 2.8.5 ,the climate efficacy can vary between different forcing agents (within 25% for most realistic RFs). Fuglestvedt et al. 2003 [JoC, MoS] ) proposed a revised GWP concept that includes the efficacy of a forcing agent. Berntsen et al. 2005 [JoC, SRC] ) calculated GWP values in this way for NOx and CO emissions in Europe and in South East Asia. The efficacies are less uncertain than climate sensitivities. However, Berntsen et al. 2005 [JoC, SRC] ) showed that for ozone produced by NOx emissions the climate efficacies will also depend on the location of the emissions.
2.10.4.2 The Global Temperature Potential
Shine et al. 2005b [JoC, SRC] ) proposed the GTP as a new relative emission metric. The GTP is defined as the ratio between the global mean surface temperature change at a given future time horizon (TH) following an emission (pulse or sustained) of a compound x relative to a reference gas r (e.g., CO2):

whereDTHx denotes the global mean surface temperature change after H years following an emission of compound x.The GTPs do not require simulations with AOGCMs, but are given as transparent and simple formulas that employ a small number of input parameters required for calculation. Note that while the GWP is an integral quantity over the time horizon (i.e., the contribution of the RF at the beginning and end of the time horizon is exactly equal), the GTP uses the temperature change at time H (i.e., RF closer to time H contributes relatively more). The GTP metric requires knowledge of the same parameters as the GWP metric (radiative efficiency and lifetimes), but in addition, the response times for the climate system must be known, in particular if the lifetime of component x is very different from the lifetime of the reference gas. Differences in climate efficacies can easily be incorporated into the GTP metric. Due to the inclusion of the response times for the climate system, the GTP values for pulse emissions of gases with shorter lifetimes than the reference gas will be lower than the corresponding GWP values. As noted by Shine et al. 2005b [JoC, SRC] ), there is a near equivalence between the GTP for sustained emission changes and the pulse GWP. The GTP metric has the potential advantage over GWP that it is more directly related to surface temperature change.
References
Abdul-Razzak, H., and S.J. Ghan, 2002: A parametrization of aerosol activation: 3. Sectional representation. J. Geophys. Res., 107(D3), 4026, doi:10.1029/2001JD000483. [JoC]
Abel, S.J., E.J. Highwood, J.M. Haywood, and M.A. Stringer, 2005: The direct radiative effect of biomass burning aerosol over southern Africa. Atmos. Chem. Phys. Discuss., 5,1165–1211. [SRC]
Abel, S.J., et al., 2003: Evolution of biomass burning aerosol properties from an agricultural fire in southern Africa. Geophys. Res. Lett., 30(15), 1783, doi:10.1029/2003GL017342. [JoC]
Ackerman, A.S, M.P. Kirkpatrick, D.E. Stevens, and O.B. Toon, 2004: The impact of humidity above stratiform clouds on indirect aerosol climate forcing. Nature, 432,1014–1017. [JoC, MoS]
Ackerman, A.S., et al., 2000a: Reduction of tropical cloudiness by soot. Science, 288,1042–1047. [JoC]
Ackerman, A.S., et al., 2000b: Effects of aerosols on cloud albedo: evaluation of Twomey’s parametrization of cloud susceptibility using measurements of ship tracks. J. Atmos. Sci., 57,2684–2695. [JoC]
Adams, P.J. and J.H. Seinfeld, 2002: Predicting global aerosol size distributions in general circulation models. J. Geophys. Res., 107(D19), 4370, doi:10.1029/2001JD001010. [JoC, MoS]
Adams, P.J., et al., 2001: General circulation model assessment of direct radiative forcing by the sulfate-nitrate-ammonium-water inorganic aerosol system. J. Geophys. Res., 106(D1), 1097–1112. [JoC, MoS]
Albrecht, B., 1989: Aerosols, cloud microphysics and fractional cloudiness. Science, 245,1227–1230. [JoC]
Allan, W., et al., 2005: Interannual variation of 13Cin tropospheric methane: Implications for a possible atomic chlorine sink in the marine boundary layer. J. Geophys. Res., 110,D11306, doi:10.1029/2004JD005650. [JoC]
Ammann, C.M., G.A. Meehl, W.M. Washington, and C.S. Zender, 2003: A monthly and latitudinally varying volcanic forcing dataset in simulations of 20th century climate. Geophys. Res. Lett., 30(12), 1657, doi:10.1029/2003GL016875. [PoC, JoC, MoS, ARC]
Anderson, T.L., et al., 2003: Climate forcing by aerosols: a hazy picture. Science, 300,1103–1104. [JoC, MoS]
Anderson, T.L., et al., 2005a. Testing the MODIS satellite retrieval of aerosol fine-mode fraction. J. Geophys. Res., 110, D18204, doi:10.1029/2005JD005978. [JoC]
Anderson, T.L., et al., 2005b: An “A-Train” strategy for quantifying direct climate forcing by anthropogenic aerosols. Bull. Am. Meteorol. Soc., 86,1795–1809. [JoC, MoS]
Andreae, M.O., and P. Merlet, 2001: Emission of trace gases and aerosols from biomass burning. Global Biogeochem. Cycles, 15(4), 955–966, doi:10.1029/2000GB001382. [JoC]
Andreae, M.O., et al., 2004: Atmospheric science: smoking rain clouds over the Amazon. Science, 303(5662), 1337–1341. [JoC]
Andres, R.J., G. Marland, T. Boden, and S. Bischof, 2000: Carbon dioxide emissions from fossil fuel consumption and cement manufacture, 1751-1991, and an estimate of their isotopic composition and latitudinal distribution. In: The Carbon Cycle [Wigley, T.M.L., and D.S. Schimel (eds.)]. Cambridge University Press, Cambridge, UK, pp. 53–62. [NPR, MoS]
Andronova, N.G., et al., 1999: Radiative forcing by volcanic aerosols from 1850 to 1994. J. Geophys. Res., 104(D14), 16807–16826. [JoC, MoS, ARC]
Antuña, J.C., et al., 2003: Spatial and temporal variability of the stratospheric aerosol cloud produced by the 1991 Mount Pinatubo eruption. J. Geophys. Res., 108,4624, doi:10.1029/2003JD003722. [JoC]
Asano, S., et al., 2002: Two case studies of winter continental-type water and mixed-phased stratocumuli over the sea. II: Absorption of solar radiation. J. Geophys. Res., 107(D21), 4570, doi:10.1029/2001JD001108. [JoC]
Baliunas, S., and R. Jastrow, 1990: Evidence for long-term brightness changes of solar-type stars. Nature, 348,520–522. [JoC]
Balkanski, Y., et al., 2004: Global emissions of mineral aerosol: formulation and validation using satellite imagery. In: Emission of Atmospheric Trace Compounds [Granier, C., P. Artaxo, and C.E. Reeves (eds.)]. Kluwer, Amsterdam, pp. 239–267. [NPR]
Bange, H.W., S. Rapsomanikis, and M.O. Andreae, 1996: Nitrous oxide in coastal waters. Global Biogeochem. Cycles, 10(1), 197–207. [JoC]
Baran, A.J., and J.S. Foot, 1994: A new application of the operational sounder HIRS in determining a climatology of sulfuric acid aerosol from the Pinatubo eruption. J. Geophys. Res., 99(D12), 25673–25679. [JoC]
Barnes, J.E., and D.J. Hoffman, 1997: Lidar measurements of stratospheric aerosol over Mauna Loa Observatory. Geophys. Res. Lett., 24(15), 1923–1926. [JoC]
Barnston, A.G., and P.T. Schickedanz, 1984: The effect of irrigation on warm season precipitation in the southern Great Plains. J. Appl. Meteorol., 23,865–888. [JoC]
Bartholome, E., and A.S. Belward, 2005: GLC2000: a new approach to global land cover mapping from Earth observation data. Int. J. Remote Sens., 26,1959–1977. Clean
Bates, T.S., et al., 2006: Aerosol direct radiative effects over the northwest Atlantic, northwest Pacific, and North Indian Oceans: estimates based on in situ chemical and optical measurements and chemical transport modelling. Atmos. Chem. Phys. Discuss., 6,175–362. [MoS]
Bauer, S.E., and D. Koch, 2005: Impact of heterogeneous sulfate formation at mineral dust surfaces on aerosol loads and radiative forcing in the Goddard Institute for Space Studies general circulation model. J. Geophys. Res., 110,D17202, doi:10.1029/2005JD005870. [JoC, MoS]
Bauman, J.J., P.B. Russell, M.A. Geller, and P. Hamill, 2003: A stratospheric aerosol climatology from SAGE II and CLAES measurements: 2. Results and comparisons, 1984-1999. J. Geophys. Res., 108(D13), 4383, doi:10.1029/2002JD002993. [JoC, SRC]
Beheng, K.D., 1994: A parametrization of warm cloud microphysical conversion process. Atmos. Res., 33,193–206. Clean
Bekki, S., K. Law, and J. Pyle, 1994: Effect of ozone depletion on atmospheric CH4 and CO concentrations. Nature, 371,595–597. [JoC]
Bellouin, N., O. Boucher, D. Tanré, and O. Dubovik, 2003: Aerosol absorption over the clear-sky oceans deduced from POLDER-1 and AERONET observations. Geophys. Res. Lett., 30(14), 1748, doi:10.1029/2003GL017121. [JoC, SRC]
Bellouin, N., O. Boucher, J. Haywood, and M.S. Reddy, 2005: Global estimates of aerosol direct radiative forcing from satellite measurements. Nature, 438,1138–1141. [JoC, MoS, SRC]
Benestad, R.E., 2005: A review of the solar cycle length estimates. Geophys. Res. Lett., 32,L15714, doi:10.1029/2005GL023621. [JoC, MoS, ARC]
Bergamaschi, P., et al., 2005: Inverse modelling of national and European CH4 emissions using the atmospheric zoom model TM5. Atmos. Chem. Phys., 5,2431–2460. [MoS]
Bergstrom, R.W., P. Pilewskie, B. Schmid, and P.B. Russell, 2003: Estimates of the spectral aerosol single scattering albedo and aerosol radiative effects during SAFARI 2000. J. Geophys. Res., 108(D13), 8474, doi:10.1029/2002JD002435. [JoC, MoS]
Berntsen, T.K., et al., 2005: Climate response to regional emissions of ozone precursors; sensitivities and warming potentials. Tellus, 4B,283–304. [JoC, SRC]
Berntsen, T.K., et al., 2006: Abatement of greenhouse gases: does location matter? Clim. Change, 74,377–411. [JoC, SRC]
Berry, E.X., 1967: Cloud droplet growth by collection. J. Atmos. Sci., 24,688–701. [JoC]
Betts, R.A., 2000: Offset of the potential carbon sink from boreal forestation by decreases in surface albedo. Nature, 408(6809), 187–189. [JoC, SRC]
Betts, R.A., 2001: Biogeophysical impacts of land use on present-day climate: near-surface temperature change and radiative forcing. Atmos. Sci. Lett., 2(1–4), doi:1006/asle.2001.0023. [MoS, SRC]
Betts, R.A., and M.J. Best, 2004: Relative impact of radiative forcing, landscape effects and local heat sources on simulated climate change in urban areas. BETWIXT Technical Briefing Note No. 6, Met Office, Exeter, UK, 15 pp. [NPR, MoS, SRC]
Betts, R.A., P.M. Cox, S.E. Lee, and F.I. Woodward, 1997: Contrasting physiological and structural vegetation feedbacks in climate change simulations. Nature, 387,796–799. [JoC, MoS, SRC]
Betts, R.A., P.D. Falloon, K.K. Goldewijk, and N. Ramankutty, 2007: Biogeophysical effects of land use on climate: model simulations of radiative forcing and large-scale temperature change. Agric. For. Meteorol., 142, 216-233. [MoS, SRC, 2007]
Betts, R.A., et al., 2004: The role of ecosystem-atmosphere interactions in simulated Amazonian precipitation decrease and forest dieback under global climate warming. Theor. Appl. Climatol., 78,157–175. [MoS, SRC]
Bigler, M., et al., 2002: Sulphate record from a northeast Greenland ice core over the last 1200 years based on continuous flow analysis. Ann.Glaciol.,35, 250–256. Clean
Bingen, C., D. Fussen, and F. Vanhellemont, 2004: A global climatology of stratospheric aerosol size distribution parameters derived from SAGE II data over the period 1984-2000: 2. Reference data. J. Geophys. Res., 109,D06202, doi:10.1029/2003JD003511. [JoC]
Blake, D., and F. Rowland, 1988: Continuing worldwide increase in tropospheric methane, 1978 to 1987. Science, 239,1129–1131. [JoC]
Blake, N.J., et al., 2001: Large-scale latitudinal and vertical distributions of NMHCs and selected halocarbons in the troposphere over the Pacific Ocean during the March-April 1999 Pacific Exploratory Mission (PEM-Tropics B). J. Geophys. Res., 106,32627–32644. [JoC]
Bluth, G.J.S., W.I. Rose, I.E. Sprod, and A.J. Krueger, 1997: Stratospheric loading of sulfur from explosive volcanic eruptions. J. Geol., 105,671–683. Clean
Bluth, G.J.S., et al., 1992: Global tracking of the SO2 clouds from the June 1991 Mount Pinatubo eruptions. Geophys. Res. Lett., 19(2), 151–154, doi:10.1029/91GL02792. [JoC]
Boberg, F., and H. Lundstedt, 2002: Solar wind variations related to fluctuations of the North Atlantic Oscillation. Geophys. Res. Lett., 29(15), doi:10.1029/2002GL014903. [JoC]
Boer, G.J., and B. Yu, 2003a: Climate sensitivity and climate state. Clim. Dyn., 21,167–176. [JoC]
Boer, G.J., and B. Yu, 2003b: Climate sensitivity and response. Clim. Dyn., 20,415–429. [JoC]
Bond, T.C., 2001: Spectral dependence of visible light absorption by carbonaceous particles emitted from coal combustion. Geophys. Res. Lett., 28(21), 4075–4078. [JoC]
Bond, T.C., and H. Sun, 2005: Can reducing black carbon emissions counteract global warming? Environ. Sci. Technol., 39,5921–5926. Clean
Bond, T.C., et al., 1999: Light absorption by primary particle emissions from a lignite burning plant. Environ. Sci.Technol., 33,3887–3891. Clean
Bond, T.C., et al., 2004: A technology-based global inventory of black and organic carbon emissions from combustion. J. Geophys. Res., 109,D14203, doi:10.1029/2003JD003697. [JoC]
Born, M., H. Dorr, and I. Levin, 1990: Methane consumption in aerated soils of the temperate zone. Tellus , 42B,2–8. [JoC]
Bortz, S.E., and M.J. Prather, 2006: Ozone, water vapor, and temperature in the upper tropical troposphere: Variations over a decade of MOZAIC Measurements. J. Geophys. Res., 111,D05305, doi:10.1029/2005JD006512. [JoC, ARC]
Boucher, O., 1999: Air traffic may increase cirrus cloudiness. Nature, 397,30–31. [JoC, SRC]
Boucher, O., and U. Lohmann, 1995: The sulfate-CCN-cloud albedo effect: a sensitivity study using two general circulation models. Tellus, 47B,281–300. [JoC, MoS, SRC]
Boucher, O., and D. Tanré, 2000: Estimation of the aerosol perturbation to the Earth’s radiative budget over oceans using POLDER satellite aerosol retrievals. Geophys. Res. Lett., 27(8), 1103–1106. [JoC, MoS, SRC]
Boucher, O., and J. Haywood, 2001: On summing the components of radiative forcing of climate change. Clim. Dyn., 18,297–302. [JoC, MoS, SRC]
Boucher, O., and M. Pham, 2002: History of sulfate aerosol radiative forcings. Geophys. Res. Lett., 29(9), 22–25. [JoC, MoS, SRC]
Boucher, O., H. Le Treut, and M.B. Baker, 1995: Precipitation and radiation modeling in a general circulation model: introduction of cloud microphysical processes. J. Geophys. Res., 100(D8), 16395–16414. [JoC, MoS, SRC]
Boucher, O., G. Myhre, and A. Myhre, 2004: Direct human influence of irrigation on atmospheric water vapour and climate. Clim. Dyn., 22,597–604. [JoC, SRC]
Bounoua, L., et al., 2002: Effects of land cover conversion on surface climate. Clim. Change, 52,29–64. [JoC]
Bousquet, P., et al., 2005: Two decades of OH variability as inferred by an inversion of atmospheric transport and chemistry of methyl chloroform. Atmos. Chem. Phys., 5,1679–1731. [ARC]
Bower, K.N., et al., 1994: A parametrization of warm clouds for use in atmospheric general circulation models. J. Atmos. Sci., 51,2722–2732. [JoC, MoS]
BP, 2006: Quantifying Energy: BP Statistical Review of World Energy June 2006.BP p.l.c., London, 45 pp., http://www.bp.com/productlanding.do?categoryId=6842&contentId=7021390. [NPR]
Bradford, D.F., 2001: Global change: time, money and tradeoffs. Nature, 410,649–650, doi:10.1038/35070707. [JoC]
Brasseur, G., and C. Granier, 1992: Mount Pinatubo aerosols, chlorofluorocarbons and ozone depletion. Science, 257,1239–1242. [JoC, ARC]
Brenguier, J.L., H. Pawlowska, and L. Schuller, 2003: Cloud microphysical and radiative properties for parametrization and satellite monitoring of the indirect effect of aerosol on climate. J. Geophys. Res., 108(D15), 8632, doi:10.1029/2002JD002682. [JoC]
Brenguier, J.L., et al., 2000a: Radiative properties of boundary layer clouds: droplet effective radius versus number concentration. J. Atmos. Sci., 57,803–821. [JoC]
Brenguier, J.L., et al., 2000b: An overview of the ACE-2 CLOUDYCOLUMN closure experiment. Tellus, 52B,815–827. [JoC]
Bréon, F-M., and M. Doutriaux-Boucher, 2005: A comparison of cloud droplet radii measured from space. IEEE Trans. Geosci. Remote. Sens., 43,1796–1805, doi:10.1109/TGRS.2005.852838. Clean
Bréon, F.-M., D. Tanré, and S. Generoso, 2002: Aerosol effect on cloud droplet size monitored from satellite. Science, 295,834–838. [JoC]
Broccoli, A.J., et al., 2003: Twentieth-century temperature and precipitation trends in ensemble climate simulations including natural and anthropogenic forcing. J. Geophys. Res., 108(D24), 4798, doi:10.1029/2003JD003812. [JoC, MoS, ARC]
Brock, C.A., et al., 2004: Particle characteristics following cloud-modified transport from Asia to North America. J. Geophys. Res., 109,1–17. [JoC]
Brovkin, V.M., et al., 2004. Role of land cover changes for atmospheric CO2 increase and climate change during the last 150 years. Global Change Biol., 10,1253–1266. [ARC]
Brovkin, V.M., et al., 2006: Biogeophysical effects of historical land cover changes simulated by six earth system models of intermediate complexity. Clim. Dyn., 26(6), 587–600. [JoC, MoS, ARC]
Cakmur, R.V., et al., 2006: Constraining the magnitude of the global dust cycle by minimizing the difference between a model and observations. J. Geophys. Res., 111,doi:10.1029/2005JD005791. [NPR, JoC, MoS]
Campbell, J.R., et al., 2003: Micropulse Lidar observations of tropospheric aerosols over northeastern South Africa during the ARREX and SAFARI-2000 dry season experiments. J. Geophys. Res., 108(D13), 8497, doi:10.1029/2002JD002563. [JoC]
Carslaw, K.S., R.G. Harrison, and J. Kirkby, 2002: Atmospheric science: Cosmic rays, clouds, and climate. Science, 298,1732–1737. [JoC]
Chapman, G.A., A.M. Cookson, and J.J. Dobias, 1996: Variations in total solar irradiance during solar cycle 22. J. Geophys. Res., 101,13541–13548. [JoC]
Chase, T.N., et al., 2000: Simulated impacts of historical land cover changes on global climate in northern winter. Clim. Dyn., 16,93–105. [JoC, MoS]
Chen, Y., and J.E. Penner, 2005: Uncertainty analysis of the first indirect aerosol effect. Atmos. Chem. Phys., 5,2935–2948. [ARC]
Chen, Y.-H., and R.G. Prinn, 2005: Atmospheric modelling of high- and low-frequency methane observations: Importance of interannually varying transport. J. Geophys. Res., 110,D10303, doi:10.1029/2004JD005542. [JoC, MoS, SRC]
Chen, Y.-H., and R.G. Prinn, 2006: Estimation of atmospheric methane emissions between 1996-2001 using a 3D global chemical transport model. J. Geophys. Res., 111,D10307, doi:10.1029/2005JD006058. [JoC, MoS, SRC]
Chiapello, I., C. Moulin, and J.M. Prospero, 2005: Understanding the long-term variability of African dust transport across the Atlantic as recorded in both Barbados surface concentrations and large-scale Total Ozone Mapping Spectrometer (TOMS) optical thickness. J. Geophys. Res., 110,D18S10, doi:10.1029/2004JD005132. [JoC]
Chipperfield, M.P., W.J. Randel, G.E. Bodeker, and P. Johnston, 2003: Global ozone: past and future. In: Scientific Assessment of Ozone Depletion: 2002 [Ennis, C.A. (ed.)]. World Meteorological Organization, Geneva, pp. 4.1–4.90. [NPR, MoS, SRC]
Chou, M.-D., P.-K. Chan, and M. Wang, 2002: Aerosol radiative forcing derived from SeaWiFS-retrieved aerosol optical properties. J. Atmos. Sci., 59,748–757. [JoC, MoS, ARC]
Christiansen, B., 1999: Radiative forcing and climate sensitivity: the ozone experience. Q. J. R. Meteorol. Soc., 125,3011–3035. [JoC, MoS]
Christopher, S.A., and J. Zhang, 2004: Cloud-free shortwave aerosol radiative effect over oceans: strategies for identifying anthropogenic forcing from Terra satellite measurements. Geophys. Res. Lett., 31,L18101, doi:10.1029/2004GL020510. [JoC, MoS]
Christopher, S.A., J. Zhang, Y.J. Kaufman, and L. Remer, 2006: Satellite-based assessment of the top of the atmosphere anthropogenic aerosol radiative forcing over cloud-free oceans. Geophys Res. Lett., 111,L15816, doi:10.1029/2005GL025535. [JoC, MoS]
Chu, D.A., et al., 2002: Validation of MODIS aerosol optical depth retrieval over land. Geophys. Res. Lett., 29(12), doi:10.1029/2001GL013205. [JoC]
Chu, D.A., et al., 2005: Evaluation of aerosol properties over ocean from Moderate Resolution Imaging Spectroradiometer (MODIS) during ACE-Asia. J. Geophys. Res., 110,D07308, doi:10.1029/2004JD005208. [JoC]
Chuang, C.C., and J.E. Penner, 1995: Effects of anthropogenic sulfate on cloud drop nucleation and optical properties. Tellus, 47B,566–577. [JoC, ARC]
Chuang, C.C., et al., 1997: An assessment of the radiative effects of anthropogenic sulfate. J. Geophys. Res., 102(D3), 3761–3778. [JoC]
Chuang, C.C., et al., 2002: Cloud susceptibility and the first aerosol indirect forcing: Sensitivity to black carbon and aerosol concentrations. J. Geophys. Res., 107(D21), 4564, doi:10.1029/2000JD000215. [JoC, MoS]
Chuang, P.Y., 2003: Measurement of the timescale of hygroscopic growth for atmospheric aerosols. J. Geophys. Res., 108(D9), 5–13. [JoC]
Chung, C.E., V. Ramanathan, D. Kim, and I.A. Podgorny, 2005: Global anthropogenic aerosol direct forcing derived from satellite and ground-based observations. J. Geophys. Res., 110,D24207, doi:10.1029/2005JD006356. [JoC, MoS, SRC]
Chung, S.H., and J.H. Seinfeld, 2002: Global distribution and climate forcing of carbonaceous aerosols. J. Geophys. Res., 107,doi:10.1029/2001JD001397. [JoC, MoS]
Chylek, P., and J. Wong, 1995: Effect of absorbing aerosols on global radiation budget. Geophys. Res. Lett., 22(8), 929–931. [JoC]
Chylek, P., B. Henderson, and M. Mishchenko, 2003: Aerosol radiative forcing and the accuracy of satellite aerosol optical depth retrieval. J. Geophys. Res., 108(D24), 4764, doi:10.1029/2003JD004044. [JoC, MoS]
Clarke, A.D., et al., 2004: Size distributions and mixtures of dust and black carbon aerosol in Asian outflow: Physiochemistry and optical properties. J. Geophys. Res., 109,D15S09, doi:10.1029/2003JD004378. [JoC]
Claquin, T., M. Schulz, and Y. Balkanski, 1999: Modeling the mineralogy of atmospheric dust. J. Geophys. Res., 104(D18), 22243–22256. [JoC, MoS, SRC]
Coen, M.C., et al., 2004: Saharan dust events at the Jungfraujoch: detection by wavelength dependence of the single scattering albedo and first climatology analysis. Atmos. Chem. Phys., 4,2465–2480. Clean
Collins, W.D., et al., 2001: Simulating aerosols using a chemical transport model with assimilation of satellite aerosol retrievals: Methodology for INDOEX. J. Geophys. Res., 106(D7), 7313–7336. [JoC, MoS, SRC]
Collins, W.D., et al., 2006: Radiative forcing by well-mixed greenhouse gases: Estimates from climate models in the IPCC AR4. J. Geophys. Res., 111,D14317, doi:10.1029/2005JD006713. [JoC, MoS, SRC]
Collins, W.J., R.G. Derwent, C.E. Johnson, and D.S. Stevenson, 2002: The oxidation of organic compounds in the troposphere and their global warming potentials. Clim. Change, 52,453–479. [JoC, ARC]
Considine, D.B., J.E. Rosenfield, and E.L. Fleming, 2001: An interactive model study of the influence of the Mount Pinatubo aerosol on stratospheric methane and water trends. J. Geophys. Res., 106(D21), 27711–27727. [JoC, MoS]
Conway, T.J., et al., 1994: Evidence for interannual variability of the carbon cycle from the NOAA/CMDL sampling network. J. Geophys. Res., 99(D11), 22831–22855. [JoC, SRC]
Cook, J., and E.J. Highwood, 2004: Climate response to tropospheric absorbing aerosols in an intermediate general-circulation model. Q. J. R. Meteorol. Soc., 130,175–191. [JoC, MoS]
Cooke, W.F., V. Ramaswamy, and P. Kasibhatla, 2002: A general circulation model study of the global carbonaceous aerosol distribution. J. Geophys. Res., 107(D16), doi:10.1029/2001JD001274. [JoC, MoS, SRC]
Cooke, W.F., C. Liousse, H. Cachier, and J. Feichter, 1999: Construction of a 1° x 1° fossil fuel emission data set for carbonaceous aerosol and implementation and radiative impact in the ECHAM4 model. J. Geophys. Res., 104(D18), 22137–22162. [JoC, MoS, ARC]
Coughlin, K., and K.K. Tung, 2004: Eleven-year solar cycle signal throughout the lower atmosphere. J. Geophys. Res., 109,D21105, doi:10.1029/2004JD004873. [JoC]
Cox, P.M., et al., 1999: The impact of new land surface physics on the GCM simulation of climate and climate sensitivity. Clim. Dyn. 15,183–203. [JoC, MoS, ARC]
Cramer, W., et al., 2001: Global response of terrestrial ecosystem structure and function to CO2 and climate change: Results from six dynamic global vegetation models. Global Change Biol., 7,357–373. [MoS]
Crooks, S.A., and L.J. Gray, 2005: Characterization of the 11-year solar signal using a multiple regression analysis of the ERA-40 dataset. J. Clim., 18,996–1015. [JoC, ARC]
Crutzen, P.J., 2004: New directions: the growing urban heat and pollution “island” effect - impact on chemistry and climate. Atmos. Environ., 38,3539–3540. Clean
Cunnold, D.M., et al., 2002: In situ measurements of atmospheric methane at GAGE/AGAGE sites during 1985-2000 and resulting source inferences. J. Geophys. Res., 107(D14), doi:10.1029/2001JD001226. [JoC]
Cunnold, D.M., et al., 2004: Comment on “Enhanced upper stratospheric ozone: Sign of recovery or solar cycle effect?” by W. Steinbrecht et al. J. Geophys. Res., 109,D14305, doi:10.1029/2004JD004826. [JoC]
Cziczo, D.J., D.M. Murphy, P.K. Hudson, and D.S. Thompson, 2004: Single particle measurements of the chemical composition of cirrus ice residue from CRYSTAL-FACE. J. Geophys. Res., 109,D04201, doi:10.1029/2003JD004032. [JoC]
Dameris, M., et al., 2005: Long-term changes and variability in a transient simulation with a chemistry-climate model employing realistic forcing. Atmos. Chem. Phys. Discuss., 5,2121–2145. [MoS, ARC]
Daniel, J.S., S. Solomon, and D.L. Albritton, 1995: On the evaluation of halocarbon radiative forcing and global warming potentials. J. Geophys. Res., 100(D1), 1271–1286, doi:10.1029/94JD02516. [PoC, JoC, MoS, ARC]
de Ridder, K., and H. Gallée, 1998: Land surface-induced regional climate change in southern Israel. J. Appl. Meteorol., 37,1470–1485. [JoC]
Decesari, S., M.C. Facchini, S. Fuzzi, and E. Tagliavini, 2000: Characterization of water-soluble organic compounds in atmospheric aerosol: a new approach. J. Geophys. Res., 105(D1), 1481–1489. [JoC, ARC]
Decesari, S., et al., 2001: Chemical features and seasonal variation of fine aerosol water-soluble organic compounds in the Po Valley, Italy. Atmos. Environ., 35,3691–3699. Clean
Del Genio, A.D., M.S. Yao, and K.-W. Lo, 1996: A prognostic cloud water parametrization for global climate models. J. Clim., 9,270–304. [JoC, MoS]
Delworth, T.L., V. Ramaswamy, and G.L. Stenchikov, 2005: The impact of aerosols on simulated ocean temperature and heat content in the 20th century. Geophys. Res. Lett., 32,L24709, doi:10.1029/2005GL024457. [JoC, MoS, SRC]
den Elzen, M., et al., 2005. Analysing countries’ contribution to climate change: Scientific and policy-related choices. Environ. Sci. Technol., 8(6), 614–636. Clean
Dentener, F., et al., 2006: Emissions of primary aerosol and precursor gases in the years 2000 and 1750 - prescribed data-sets for AeroCom. Atmos. Chem. Phys. Discuss., 6,2703–2763. [ARC]
Derwent, R.G., W.J. Collins, C.E. Johnson, and D.S. Stevenson, 2001: Transient behaviour of tropospheric ozone precursors in a global 3-D CTM and their indirect greenhouse effects. Clim. Change, 49,463–487. [JoC, ARC]
Deshler, T., et al., 2003: Thirty years of in situ stratospheric aerosol size distribution measurements from Laramie, Wyoming (41N), using balloon-borne instruments. J. Geophys. Res., 108(D5), 4167, doi:10.1029/2002JD002514. [JoC]
Deuzé, J.L., et al., 2000: Estimate of the aerosol properties over the ocean with POLDER. J. Geophys. Res., 105,15329–15346. [JoC, MoS]
Deuzé, J.L., et al., 2001: Remote sensing of aerosols over land surfaces from POLDER-ADEOS-1 polarized measurements. J. Geophys. Res., 106,4913–4926. [JoC]
Dewitte, S., D. Crommelynck, S. Mekaoui, and A. Joukoff, 2005: Measurement and uncertainty of the long-term total solar irradiance trend. Sol. Phys., 224,209–216. Clean
Diab, R.D., et al., 2004: Tropospheric ozone climatology over Irene, South Africa, from 1990 to 1994 and 1998 to 2002. J. Geophys. Res., 109,D20301, doi:10.1029/2004JD004793. [JoC]
Dlugokencky, E.J., K.A. Masarie, P.M. Lang, and P.P. Tans, 1998: Continuing decline in the growth rate of atmospheric methane. Nature, 393,447–450. [JoC, SRC]
Dlugokencky, E.J., et al., 1996: Changes in CH4 and CO growth rates after the eruption of Mt. Pinatubo and their link with changes in tropical tropospheric UV flux. Geophys. Res. Lett., 23(20), 2761–2764. [JoC, SRC]
Dlugokencky, E.J., et al., 2001: Measurements of an anomalous global methane increase during 1998. Geophys. Res. Lett., 28(3), 499–502. [JoC, SRC]
Dlugokencky, E.J., et al., 2003: Atmospheric methane levels off: Temporary pause or a new steady-state? Geophys. Res. Lett., 30(19), doi:10.1029/2003GL018126. [JoC, SRC]
Dlugokencky, E.J., et al., 2005: Conversion of NOAA CMDL atmospheric dry air CH4 mole fractions to a gravimetrically prepared standard scale. J. Geophys. Res., 110,D18306, doi:10.1029/2005JD006035. [JoC, SRC]
Döll, P., 2002: Impact of climate change and variability on irrigation requirements: a global perspective. Clim. Change, 54,269–293. [JoC]
Douglass, A., M. Schoeberl, R. Rood, and S. Pawson, 2003: Evaluation of transport in the lower tropical stratosphere in a global chemistry and transport model. J. Geophys. Res., 108(D9), 4259, doi:10.1029/2002JD002696. [JoC, MoS]
Douglass, D.H., and B.D. Clader, 2002: Climate sensitivity of the Earth to solar irradiance. Geophys. Res. Lett., 29(16), 33–36. [JoC]
Dubovik, O., and M.D. King, 2000: A flexible inversion algorithm for retrieval of aerosol optical properties from Sun and sky radiance measurements. J. Geophys. Res., 105,20673–20696. [JoC]
Dubovik, O., et al., 1998: Single-scattering albedo of smoke retrieved from the sky radiance and solar transmittance measured from ground. J. Geophys. Res., 103(D24), 31903–31923. [JoC]
Dubovik, O., et al., 2000: Accuracy assessments of aerosol optical properties retrieved from Aerosol Robotic Network (AERONET) Sun and sky radiance measurements. J. Geophys. Res., 105,9791–9806. [JoC]
Dubovik, O., et al., 2002: Variability of absorption and optical properties of key aerosol types observed in worldwide locations. J. Atmos. Sci., 59,590–608. [JoC]
Dufresne, L.-L., et al., 2005: Contrasts in the effects on climate of anthropogenic sulfate aerosols between the 20th and the 21st century. Geophys. Res. Lett., 32,L21703, doi:10.1029/2005GL023619. [JoC]
Duncan, B.N., et al., 2003: Indonesian wildfires of 1997: Impact on tropospheric chemistry. J. Geophys. Res., 108(D15), 4458, doi:10.1029/2002JD003195. [JoC]
Dusek, U., et al., 2006: Size matters more than chemistry for cloud-nucleating ability of aerosol particles. Science, 312,1375–1378. [JoC]
Dziembowski, W.A., P.R. Goode, and J. Schou, 2001: Does the sun shrink with increasing magnetic activity? Astrophys.J., 553,897–904. Clean
Eagan, R., P.V. Hobbs, and L. Radke, 1974: Measurements of CCN and cloud droplet size distributions in the vicinity of forest fires. J. Appl. Meteorol., 13,537–553. [JoC]
Easter, R., et al., 2004: MIRAGE: Model description and evaluation of aerosols and trace gases. J. Geophys. Res. 109,D20210, doi:10.1029/2004JD004571. [JoC, MoS]
Eatough, D.J., et al., 2003: Semivolatile particulate organic material in southern Africa during SAFARI 2000. J. Geophys. Res., 108(D13), 8479, doi:10.1029/2002JD002296. [JoC]
Eck, T.F., et al., 2003: Variability of biomass burning aerosol optical characteristics in southern Africa during the SAFARI 2000 dry season campaign and a comparison of single scattering albedo estimates from radiometric measurements. J. Geophys. Res., 108(D13), 8477, doi:10.1029/2002JD002321. [JoC, MoS]
Edwards, D.P., et al., 2004: Observations of carbon monoxide and aerosols from the Terra satellite: Northern Hemisphere variability. J. Geophys. Res., 109,D24202, doi:10.1029/2004JD004727. [JoC]
Ehhalt, D.H., 1999: Gas phase chemistry of the troposphere. In: Global Aspects of Atmospheric Chemistry, Vol. 6 [Baumgaertel, H., W. Gruenbein, and F. Hensel (eds.)]. Springer Verlag, Darmstadt, pp. 21–110. [NPR]
EPA, 2003: National Air Quality and Emissions Trends Report, 2003 Special Studies Edition.Publication No. EPA 454/R-03-005, U. S. Environmental Protection Agency, Washington, DC, 190pp, http://www.epa.gov/air/airtrends/aqtrnd03/ [NPR]
Erlick, C., L.M. Russell, and V. Ramaswamy, 2001: A microphysics-based investigation of the radiative effects of aerosol-cloud interactions for two MAST Experiment case studies. J. Geophys. Res., 106(D1), 1249–1269. [JoC, SRC]
Ervens, B., G. Feingold, and S.M. Kreidenweis, 2005: The influence of water-soluble organic carbon on cloud drop number concentration. J. Geophys. Res., 110,D18211, doi:10.1029/2004JD005634. [JoC]
Etheridge, D.M., L.P. Steele, R.J. Francey, and R.L. Langenfelds, 1998: Atmospheric methane between 1000 A.D. and present: Evidence of anthropogenic emissions and climatic variability. J. Geophys. Res., 103(D13), 15979–15993. [JoC, SRC]
Etheridge, D.M., et al., 1996: Natural and anthropogenic changes in atmospheric CO2 over the last 1000 years from air in Antarctic ice and firn. J. Geophys. Res., 101(D2), 4115–4128. [JoC, SRC]
Facchini, M.C., M. Mircea, S. Fuzzi, and R.J. Charlson, 1999: Cloud albedo enhancement by surface-active organic solutes in growing droplets. Nature, 401,257–259. [JoC, ARC]
Facchini, M.C., et al., 2000: Surface tension of atmospheric wet aerosol and cloud/fog droplets in relation to their organic carbon content and chemical composition. Atmos. Environ., 34,4853–4857. Clean
Feichter, J., E. Roeckner, U. Lohmann, and B. Liepert, 2004: Nonlinear aspects of the climate response to greenhouse gas and aerosol forcing. J. Clim., 17,2384–2398. [JoC, MoS, SRC]
Feingold, G., 2003: Modelling of the first indirect effect: Analysis of measurement requirements. Geophys. Res. Lett., 30(19), 1997, doi:10.1029/2003GL017967. [JoC, MoS]
Feingold, G., and P.Y. Chuang, 2002: Analysis of influence of film-forming compounds on droplet growth: Implications for cloud microphysical processes and climate. J. Atmos. Sci., 59,2006–2018. [JoC]
Feingold, G., R. Boers, B. Stevens, and W.R. Cotton, 1997: A modelling study of the effect of drizzle on cloud optical depth and susceptibility.J. Geophys. Res., 102(D12), 13527–13534. [JoC, MoS]
Feingold, G., W.L. Eberhard, D.E. Veron, and M. Previdi, 2003: First measurements of the Twomey indirect effect using ground-based remote sensors. Geophys. Res. Lett., 30(6), 1287, doi:10.1029/2002GL016633. [JoC]
Feng, Y., and J. Penner. 2007: Global modeling of nitrate and ammonium: Interaction of aerosols and tropospheric chemistry. J. Geophys. Res., 112(D01304), doi:10.1029/2005JD006404. [JoC, MoS, ARC, 2007]
Ferretti, D.F., D.C. Lowe, R.J. Martin, and G.W. Brailsford, 2000: A new GC-IRMS technique for high precision,N2O-free analysis of δ13Cand δ18Oin atmospheric CO2 from small air samples. J. Geophys. Res., 105(D5), 6709–6718. [JoC, SRC]
Field, C.B., R.B. Jackson, and H.A. Mooney, 1995: Stomatal responses to increased CO2:implications from the plant to the global scale. Plant Cell Environ., 18,1214–1225. Clean
Fioletov, V.E., et al., 2002: Global and zonal total ozone variations estimated from ground-based and satellite measurements: 1964-2000. J. Geophys. Res., 107(D22), 4647, doi:10.1029/2001JD001350. [JoC, MoS]
Formenti, P., et al., 2003: Inorganic and carbonaceous aerosols during the Southern African Regional Science Initiative (SAFARI 2000) experiment: Chemical characteristics, physical properties, and emission data or smoke from African biomass burning. J. Geophys. Res., 108(D13), 8488, doi:10.1029/2002JD002408. [JoC]
Forster, P.M.F., and K.P. Shine, 1999: Stratospheric water vapour changes as a possible contributor to observed stratospheric cooling. Geophys. Res. Lett., 26(21), 3309–3312. [JoC, SRC]
Forster, P.M.F., and K. Tourpali, 2001: Effect of tropopause height changes on the calculation of ozone trends and their radiative forcing. J. Geophys. Res., 106(D11), 12241–12251. [JoC, MoS, SRC]
Forster, P.M.F., and K.P. Shine, 2002: Assessing the climate impact of trends in stratospheric water vapor. Geophys. Res. Lett., 29(6), doi:10.1029/2001GL013909. [JoC, SRC]
Forster, P.M.F., and M.J. Joshi, 2005: The role of halocarbons in the climate change of the troposphere and stratosphere. Clim. Change, 70,249–266. [JoC, SRC]
Forster, P.M.F., and K.E. Taylor, 2006: Climate forcings and climate sensitivities diagnosed from coupled climate model integrations. J. Clim., 19,6181–6194. [JoC, MoS, SRC]
Forster, P.M.F., M. Blackburn, R. Glover, and K.P. Shine, 2000: An examination of climate sensitivity for idealised climate change experiments in an intermediate general circulation model. Clim. Dyn., 16,833–849. [JoC, MoS, SRC]
Forster, P.M.F., et al., 2005: Resolving uncertainties in the radiative forcing of HFC-134s. J. Quant. Spectrosc. Radiative Transfer, 93,447–460. [MoS, SRC]
Foster, S.S., 2004: Reconstruction of Solar Irradiance Variations for use in Studies of Global Climate Change: Application of Recent SOHO Observations with Historic Data from the Greenwich Observatory. PhD Thesis, University of Southampton, Faculty of Science, Southampton, 231 p. [NPR]
Foukal, P., C. Frohlich, H. Spruit, and T.M.L. Wigley, 2006: Variations in solar luminosity and their effect on the Earth’s climate. Nature, 443,161–166. [PoC, JoC, SRC]
Francey, R.J., et al., 1995: Changes in oceanic and terrestrial carbon uptake since 1982. Nature, 373,326–330. [JoC]
Francey, R.J., et al., 1999: A history of δ13Cin atmospheric CH4 from the Cape Grim Air Archive and Antarctic firn air.J. Geophys. Res., 104(D19), 23633–23643. [JoC]
Frankenberg, C., et al., 2005: Assessing methane emissions from global space-borne observations. Science, 308,1010–1014. [JoC]
Free, M., and J. Angell, 2002: Effect of volcanoes on the vertical temperature profile in radiosonde data.J. Geophys. Res., 107(D10), doi:10.1029/2001JD001128. [JoC, ARC]
Friedl, M.A., et al., 2002: Global land cover mapping from MODIS: algorithms and early results. Remote Sens. Environ., 83,287–302. Clean
Fröhlich, C., and J. Lean, 2004: Solar radiative output and its variability: Evidence and mechanisms. Astron. Astrophys. Rev., 12,273–320. [SRC]
Fueglistaler, S., H. Wernli, and T. Peter, 2004: Tropical troposphere-to-stratosphere transport inferred from trajectory calculations. J. Geophys. Res., 109,D03108, doi:10.1029/2003JD004069. [JoC]
Fuglestvedt, J.S., T.K. Berntsen, O. Godal, and T. Skodvin, 2000: Climate implications of GWP-based reductions in greenhouse gas emissions. Geophys. Res. Lett., 27(3), 409–412, doi:10.1029/1999GL010939. [JoC, SRC]
Fuglestvedt, J.S., et al., 1999: Climatic forcing of nitrogen oxides through changes in tropospheric ozone and methane; global 3D model studies. Atmos. Environ., 33,961–977. [MoS]
Fuglestvedt, J.S., et al., 2003: Metrics of climate change: assessing radiative forcing and emission indices. Clim. Change, 58,267–331. [JoC, MoS]
Fuller, K.A., W.C. Malm, and S.M. Kreidenweis, 1999: Effects of mixing on extinction by carbonaceous particles. J. Geophys. Res., 104(D13), 15941–15954. [JoC]
Fuzzi, S., et al., 2001; A simplified model of the water soluble organic component of atmospheric aerosols. Geophys. Res. Lett., 28(21), 4079–4082. [JoC, MoS, ARC]
Gao, F., et al., 2005: MODIS bidirectional reflectance distribution function and albedo Climate Modeling Grid products and the variability of albedo for major global vegetation types. J. Geophys. Res., 110,D01104, doi:10.1029/2004JD005190. [JoC, MoS]
Gauss, M., et al., 2006: Radiative forcing since preindustrial times due to ozone changes in the troposphere and the lower stratosphere. Atmos. Chem. Phys., 6,575–599. [MoS]
Gedney, N., et al., 2006: Detection of a direct carbon dioxide effect in continental river runoff records. Nature, 439,835–838, doi:10.1038/nature04504. [JoC]
Geller, M.A., and S.P. Smyshlyaev, 2002: A model study of total ozone evolution 1979-2000: The role of individual natural and anthropogenic effects. Geophys. Res. Lett., 29(22), 5–8. [JoC, MoS, SRC]
Geogdzhayev, I.V., et al., 2002: Global two-channel AVHRR retrievals of aerosol properties over the ocean for the period of NOAA-9 observations and preliminary retrievals using NOAA-7 and NOAA-11 data. J. Atmos. Sci., 59,262–278. [JoC]
Ghan, S., et al., 2001: Evaluation of aerosol direct radiative forcing in MIRAGE. J. Geophys. Res., 106(D6), 5295–5316. [JoC, MoS]
Gillett, N.P., M.F. Wehner, S.F.B. Tett, and A.J. Weaver, 2004: Testing the linearity of the response to combined greenhouse gas and sulfate aerosol forcing. Geophys. Res. Lett., 31,L14201, doi:10.1029/2004GL020111. [JoC, MoS, ARC]
Gleisner, H., and P. Thejll, 2003: Patterns of tropospheric response to solar variability. Geophys. Res. Lett., 30(13), 1711, doi:10.1029/2003GL017129. [JoC]
Godal, O., 2003: The IPCC assessment of multidisciplinary issues: The choice of greenhouse gas indices. Clim. Change, 58,243–249. [JoC]
Gohar, L.K., G. Myhre, and K.P. Shine, 2004: Updated radiative forcing estimates of four halocarbons. J. Geophys. Res., 109,D01107, doi:10.1029/2003JD004320. [JoC, MoS, SRC]
Goloub, P., and O. Arino, 2000: Verification of the consistency of POLDER aerosol index over land with ATSR-2/ERS-2 fire product. Geophys. Res. Lett., 27(6), 899–902. [JoC]
Goloub, P., et al., 1999: Validation of the first algorithm applied for deriving the aerosol properties over the ocean using the POLDER/ADEOS measurements. IEEE Trans. Remote Sens., 37,1586–1596. Clean
Goosse, H., H. Renssen, A. Timmermann, and R.S. Bradley, 2005: Internal and forced climate variability during the last millennium: a model-data comparison using ensemble simulations. Quat. Sci. Rev., 24, 1345–1360. [PoC, JoC, MoS, ARC]
Gordon, L.J., et al., 2005: Human modification of global water vapor flows from the land surface. Proc. Natl. Acad. Sci. U.S.A., 102,7612–7617. [JoC]
Govindasamy, B., P.B. Duffy, and K. Caldeira, 2001a: Land use changes and Northern Hemisphere cooling. Geophys. Res. Lett., 28(2), 291–294. [JoC]
Govindasamy, B., et al., 2001b: Limitations of the equivalent CO2 approximation in climate change simulations. J. Geophys. Res., 106(D19), 22593–22603. [JoC, MoS]
Gray, L.J., J.D. Haigh, and R.G. Harrison, 2005: Review of the Influences of Solar Changes on the Earth’s Climate. Hadley Centre Technical Note No. 62, Met Office, Exeter, 82 pp. [NPR, ARC]
Gregory, J.M., et al., 2004: A new method for diagnosing radiative forcing and climate sensitivity. Geophys. Res. Lett., 31,L03205, doi:10.1029/2003GL018747. [JoC, MoS, ARC]
Gunn, R., and B.B. Phillips, 1957: An experimental investigation of the effect of air pollution on the initiation of rain. J. Meteorol., 14(3), 272–280. Clean
Hadjinicolaou, P., J.A. Pyle, and N.R.P. Harris, 2005: The recent turnaround in stratospheric ozone over northern middle latitudes: a dynamical modeling perspective. Geophys. Res. Lett., 32,L12821, doi:10.1029/2005GL022476. [JoC, MoS]
Haigh, J.D., 2003: The effects of solar variability on the Earth’s climate. Phil. Trans. R. Soc. London Ser.A, 361,95–111. [JoC, ARC]
Hall, J.C., and G.W. Lockwood, 2004: The chromospheric activity and variability of cycling and flat activity solar-analog stars. Astrophys. J., 614,942–946. Clean
Hamilton, J.F., et al., 2004: Partially oxidised organic components in urban aerosol using GCXGC-TOF/MS. Atmos. Chem. Phys., 4,1279–1290. Clean
Hammitt, J.K., A.K. Jain, J.L. Adams, and D.J. Wuebbles, 1996: A welfare-based index for assessing environmental effects of greenhouse-gas emissions. Nature, 381,301–303. [JoC]
Han, Q., W.B. Rossow, J. Zeng, and R. Welch, 2002: Three different behaviors of liquid water path of water clouds in aerosol-cloud interactions. J. Atmos. Sci., 59,726–735. [JoC]
Hansen, J., and M. Sato, 2001: Trends of measured climate forcing agents. Proc. Natl. Acad. Sci. U.S.A., 98,14778–14783. [JoC, MoS, ARC]
Hansen, J., and L. Nazarenko, 2004: Soot climate forcing via snow and ice albedos. Proc. Natl. Acad. Sci. U.S.A., 101,423–428. [JoC, MoS, ARC]
Hansen, J., and M. Sato, 2004: Greenhouse gas growth rates. Proc. Natl. Acad. Sci. U.S.A., 101,16109–16114. [JoC, ARC]
Hansen, J., M. Sato, and R. Reudy, 1995: Long-term changes of the diurnal temperature cycle: Implications about mechanisms of global climate change. Atmos. Res., 37,175–209. [ARC]
Hansen, J., M. Sato, and R. Ruedy, 1997: Radiative forcing and climate response. J. Geophys. Res., 102(D6), 6831–6864. [JoC, MoS, ARC]
Hansen, J., et al., 2000: Global warming in the twenty-first century: An alternative scenario. Proc. Natl. Acad. Sci. U.S.A., 97,9875–9880. [JoC, ARC]
Hansen, J., et al., 2002: Climate forcings in Goddard Institute for Space Studies SI2000 simulations. J. Geophys. Res., 107(D18), 4347, doi:10.1029/2001JD001143. [JoC, MoS, ARC]
Hansen, J., et al., 2005: Efficacy of climate forcings. J. Geophys. Res., 110,D18104, doi:10.1029/2005JD005776. [JoC, MoS, ARC]
Hansen, M.C., and B. Reed, 2000: A comparison of the IGBP DISCover and University of Maryland 1km global land cover products. Int. J. Remote Sens., 21,1365–1373. Clean
Harder, J., et al., 2005: The spectral irradiance monitor I. Scientific requirements, instrument design and operation modes. Sol. Phys., 230,141–167. Clean
Harding, R.J., and J.W. Pomeroy, 1996: The energy balance of the winter boreal landscape. J. Clim., 9,2778–2787. [JoC]
Harnisch, J., R.R. Borchers, P.P. Fabian, and M.M. Maiss, 1996: Tropospheric trends for CF4 andC2F6 since 1982 derived from SF6 dated stratospheric air. Geophys. Res. Lett., 23(10), 1099–1102. [JoC]
Harries, J.E., H.E. Brindley, P.J. Sagoo, and R.J. Bantges, 2001: Increases in greenhouse forcing inferred from the outgoing longwave radiation spectra of the Earth in 1970 and 1997. Nature, 410,355–357. [JoC, MoS]
Harrison, R.G., and K.S. Carslaw, 2003: Ion-aerosol-cloud processes in the lower atmosphere. Rev. Geophys., 41,1021. [JoC]
Harrison, R.G., and D.B. Stephenson, 2006: Empirical evidence for a nonlinear effect of galactic cosmic rays on clouds. Proc. Roy. Soc. London Ser. A, 462,1221–1233. Clean
Hartmann, D.L., M.E. Ockert-Bell, and M.L. Michelsen, 1992: The effect of cloud type on Earth’s energy balance: global analysis. J. Clim., 5, 1281–1304. [JoC]
Hashvardhan, S.E. Schwartz, C.E. Benkovitz, and G. Guo, 2002: Aerosol influence on cloud microphysics examined by satellite measurements and chemical transport modelling. J. Atmos. Sci., 59,714–725. [JoC, MoS]
Hauglustaine, D.A., and G.P. Brasseur, 2001: Evolution of tropospheric ozone under anthropogenic activities and associated radiative forcing of climate. J. Geophys. Res., 106(D23), 32337–32360, doi:10.1029/2001JD900175. [JoC, MoS, ARC]
Haxeltine, A., and I.C. Prentice, 1996: BIOME3: An equilibrium terrestrial biosphere model based on ecophysiological constraints, resource availability, and competition among plant functional types. Global Biogeochem. Cycles, 10(4), 693–709. [JoC, MoS]
Haywood, J.M., and K.P. Shine, 1995: The effect of anthropogenic sulfate and soot aerosol on the clear sky planetary radiation budget. Geophys. Res. Lett., 22(5), 603–606. [JoC, SRC]
Haywood, J.M., and K.P. Shine. 1997: Multi-spectral calculations of the direct radiative forcing of tropospheric sulphate and soot aerosols using a column model.Q. J. R. Meteorol. Soc. 123,1907–1930. [MoS, SRC]
Haywood, J.M., and O. Boucher, 2000: Estimates of the direct and indirect radiative forcing due to tropospheric aerosols: A review. Rev. Geophys., 38,513–543. [JoC, MoS, SRC]
Haywood, J.M., V. Ramaswamy, and B.J. Soden, 1999: Tropospheric aerosol climate forcing in clear-sky satellite observations over the oceans. Science, 283,1299–1305. [JoC, MoS, SRC]
Haywood, J.M., et al., 2003a: Comparison of aerosol size distributions, radiative properties, and optical depths determined by aircraft observations and Sun photometers during SAFARI 2000. J. Geophys. Res., 108(D13), 8471, doi:10.1029/2002JD002250. [JoC, SRC]
Haywood, J.M., et al., 2003b: The mean physical and optical properties of regional haze dominated by biomass burning aerosol measured from the C-130 aircraft during SAFARI 2000. J. Geophys. Res., 108(D13), 8473, doi:10.1029/2002JD002226. [JoC, SRC]
Haywood, J.M, et al., 2003c: Radiative properties and direct radiative effect of Saharan dust measured by the C-130 aircraft during SHADE: 1. Solar spectrum. J. Geophys. Res., 108(D18), 8577, doi:10.1029/2002JD002687. [JoC, SRC]
Haywood, J.M., et al., 2005: Can desert dust explain the outgoing longwave radiation anomaly over the Sahara during July 2003? J. Geophys. Res., 110,D05105, doi:10.1029/2004JD005232. [JoC, SRC]
Heal, G., 1997: Discounting and climate change: an editorial comment. Clim. Change, 37,335–343. [JoC]
Hegg, D.A., et al., 1997: Chemical apportionment of aerosol column optical depth off the mid-Atlantic coast of the United States. J. Geophys. Res., 102(D21), 25293–25303. [JoC]
Hély, C., et al., 2003: Release of gaseous and particulate carbonaceous compound from biomass burning during the SAFARI 2000 dry season field campaign. J. Geophys. Res., 108(D13), 8470, doi:10.1029/2002JD002482. [JoC]
Hendricks, J., B. Kärcher, U. Lohmann, and M. Ponater, 2005: Do aircraft black carbon emissions affect cirrus clouds on the global scale? Geophys. Res. Lett., 32,L12814, doi:10.1029/2005GL022740. [JoC, SRC]
Hendricks, J., et al., 2004: Simulating the global atmospheric black carbon cycle: a revisit to the contribution of aircraft. Atmos. Chem. Phys., 4,2521–2541. [MoS]
Herman, M., et al., 1997: Remote sensing of aerosols over land surfaces including polarization measurements and application to POLDER measurements. J. Geophys. Res., 102(D14), 17039–17050, doi:10.1029/96JD02109. [JoC]
Hess, M., P. Koepke, and I. Schult, 1998: Optical properties of aerosols and clouds: the software package OPAC. Bull. Am. Meteorol. Soc., 79,831–844. [JoC]
Highwood, E.J., et al., 2003: Radiative properties and direct effect of Saharan dust measured by the C-130 aircraft during Saharan Dust Experiment (SHADE): 2. Terrestrial spectrum. J. Geophys. Res., 108(D18), 8577, doi:10.1029/2002JD002552. [JoC]
Higurashi, A., and T. Nakajima. 1999. Development of a two-channel aerosol retrieval algorithm on a global scale using NOAA AVHRR. J. Atmos. Sci., 56, 924–941. [JoC, SRC]
Higurashi, A., et al., 2000: A study of global aerosol optical climatology with two-channel AVHRR remote sensing. J. Clim., 13,2011–2027. [JoC]
Hirsch, A.I., et al., 2006: Inverse modeling estimates of the global nitrous oxide surface flux from 1998–2001. Global Biogeochem. Cycles, 20,GB1008, doi:10.1029/2004GB002443. [JoC, MoS]
Hirst, E., et al., 2001: Discrimination of micrometre-sized ice and super-cooled droplets in mixed-phase cloud. Atmos. Environ., 35,33–47. Clean
Hofmann, D., 2003: et al., Surface-based observations of volcanic emissions to stratosphere. In: Volcanism and the Earth’s Atmosphere [Robock, A., and C. Oppenheimer (eds.)]. Geophysical Monograph 139, American Geophysical Union, Washington, DC, pp. 57–73. [NPR]
Holben, B.N., et al., 1998: AERONET: A federated instrument network and data archive for aerosol characterization. Remote Sens. Environ., 66,1–16. Clean
Holben, B.N., et al., 2001: An emerging ground-based aerosol climatology: aerosol optical depth from AERONET. J. Geophys. Res., 106(D11), 12067–12097. [JoC]
Holzer-Popp, T., M. Schroedter, and G. Gesell, 2002: Retrieving aerosol optical depth and type in the boundary layer over land and ocean from simultaneous GOME spectrometer and ATSR-2 radiometer measurements - 2. Case study application and validation. J. Geophys. Res., 107(D21), 4578, doi:10.1029/2002JD002777. [JoC, MoS]
Hood, L.L., 2003: Thermal response of the tropical tropopause region to solar ultraviolet variations. Geophys. Res. Lett., 30,2215, doi:10.1029/2003GL018364. [JoC]
Houghton, R.A., 1999: The annual net flux of carbon to the atmosphere from changes in land use 1850-1990. Tellus, 51B,298–313. [JoC]
Houghton, R.A., 2000: A new estimate of global sources and sinks of carbon from land use change. Eos, 81(19), S281. [JoC, MoS]
Houghton, R.A., 2003: Revised estimates of the annual net flux of carbon to the atmosphere from changes in land use and land management. Tellus, 55B,378–390. [JoC, MoS]
Houghton, R.A., et al., 1983: Changes in the carbon content of terrestrial biota and soils between 1860 and 1980: a net release of CO2 to the atmosphere. Ecol. Monogr., 53,235–262, doi:10.2307/1942531. Clean
Hoyt, D.V., and K.H. Schatten, 1993: A discussion of plausible solar irradiance variations, 1700-1992. J. Geophys. Res., 98(A11), 18895–18906. [JoC]
Hsu, N.C., J.R. Herman, and C. Weaver, 2000: Determination of radiative forcing of Saharan dust using combined TOMS and ERBE data.J. Geophys. Res., 105(D16), 20649–20661. [JoC, MoS]
Hsu, N.C., J.R. Herman, and S.C. Tsay, 2003: Radiative impacts from biomass burning in the presence of clouds during boreal spring in southeast Asia. Geophys. Res. Lett., 30(5), 28, doi:10.1029/2002GL016485. [JoC]
Huang, J., and R.G. Prinn, 2002: Critical evaluation of emissions of potential new gases for OH estimation. J. Geophys. Res., 107(D24), 4784, doi:10.1029/2002JD002394. [JoC, MoS, SRC]
Huebert, B.J., et al., 2003: An overview of ACE-Asia: Strategies for quantifying the relationships between Asian aerosols and their climatic impacts. J. Geophys. Res., 108(D23), 8633, doi:10.1029/2003JD003550. [JoC]
Hurley, M.D., et al., 2005: IR spectrum and radiative forcing of CF4 revisited. J. Geophys. Res., 110,D02102, doi:10.1029/2004JD005201. [JoC, MoS]
Hurst, D.F., et al., 2004: Emissions of ozone-depleting substances in Russia during 2001. J. Geophys. Res., 109,D14303, doi:10.1029/2004JD004633. [JoC]
Husar, R.B., J.M. Prospero, and L.L. Stowe, 1997: Characterization of tropospheric aerosols over the oceans with the NOAA advanced very high resolution radiometer optical thickness operational product. J. Geophys. Res., 102(D14), 16889–16910. [JoC]
Husar, R.B., et al., 2001: Asian dust events of April 1998. J. Geophys. Res., 106(D16), 18317–18330. [JoC]
Ichinose, T., K. Shimodozono, and K. Hanaki, 1999: Impact of anthropogenic heat on urban climate in Tokyo. Atmos. Environ., 33,3897–3909. Clean
Ichoku, C., et al., 2003: MODIS observation of aerosols and estimation of aerosol radiative forcing over southern Africa during SAFARI 2000. J. Geophys. Res., 108(D13), 8499, doi:10.1029/2002JD002366. [JoC, MoS]
Ignatov, A., and L. Stowe, 2002: Aerosol retrievals from individual AVHRR channels. Part I: Retrieval algorithm and transition from Dave to 6S Radiative Transfer Model. J. Atmos. Sci., 59,313–334. [JoC, MoS]
IPCC, 1990: Climate Change: The Intergovernmental Panel on Climate Change Scientific Assessment [Houghton, J.T., G.J. Jenkins, and J.J. Ephraums (eds.)]. Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, 364 pp. [NPR]
IPCC, 1999: Aviation and the Global Atmosphere: A Special Report of IPCC Working Groups I and III [Penner, J.E., et al. (eds.)]. Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, 373 pp. [NPR]
IPCC, 2001: Climate Change 2001: The Scientific Basis. Contribution of Working Group I to the Third Assessment Report of the Intergovernmental Panel on Climate Change [Houghton, J.T., et al. (eds.)]. Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, 881 pp. [NPR]
IPCC/TEAP, 2005: Special Report on Safeguarding the Ozone Layer and the Global Climate System: Issues Related to Hydrofluorocarbons and Perfluorocarbons [Metz, B., et al. (eds.)]. Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, 488 pp. [NPR]
Ito, A., and J.E. Penner, 2005: Historical emissions of carbonaceous aerosols from biomass and fossil fuel burning for the period 1870–2000. Global Biogeochem. Cycles, 19,GB2028, doi:10.1029/2004GB002374. [JoC, ARC]
Iversen, T., and O. Seland, 2002: A scheme for process-tagged SO4 and BC aerosols in NCAR CCM3: Validation and sensitivity to cloud processes. J. Geophys. Res., 107(D24), 4751, doi:10.1029/2001JD000885. [JoC]
Jacob, D.J., et al., 2005: Radiative Forcing of Climate Change.The National Academies Press, Washington, DC, 207 pp. [NPR, MoS, ARC]
Jacobson, M.Z., 1999: Isolating nitrated and aromatic aerosols and nitrated aromatic gases as sources of ultraviolet light absorption. J. Geophys. Res., 104(D3), 3527–3542. [JoC]
Jacobson, M.Z., 2001a: Global direct radiative forcing due to multicomponent anthropogenic and natural aerosols. J. Geophys. Res., 106(D2), 1551–1568. [JoC, MoS]
Jacobson, M.Z., 2001b: Strong radiative heating due to the mixing state of black carbon in atmospheric aerosols. Nature, 409,695–697. [JoC]
Jacobson, M.Z., 2002: Control of fossil-fuel particulate black carbon and organic matter, possibly the most effective method of slowing global warming. J. Geophys. Res., 107(D19), 4410, doi:10.1029/2001JD001376. [JoC]
Jacobson, M.Z., 2004: Climate response of fossil fuel and biofuel soot, accounting for soot’s feedback to snow and sea ice albedo and emissivity. J. Geophys. Res., 109,D21201, doi:10.1029/2004JD004945. [JoC]
Jaffe, D., et al., 2003: Increasing background ozone during spring on the west coast of North America. Geophys. Res. Lett., 30,1613, doi:10.1029/2003GL017024. [JoC]
Jensen, E.J., and O.B. Toon, 1997: The potential impact of soot particles from aircraft exhaust on cirrus clouds. Geophys. Res. Lett., 24(3), 249–252. [JoC]
Jeong, M.J., Z.Q. Li, D.A. Chu, and S.C. Tsay, 2005: Quality and compatibility analyses of global aerosol products derived from the advanced very high resolution radiometer and Moderate Resolution Imaging Spectroradiometer. J. Geophys. Res., 110,D10S09, doi:10.1029/2004JD004648. [JoC]
Jiang, H., and G. Feingold, 2006: Effect of aerosol on warm convective clouds: Aerosol-clouds-surface flux feedbacks in a new coupled large eddy model. J. Geophys. Res., 111,D01202, doi:10.1029/2005JD006138. [JoC, MoS]
Jiang, H., G. Feingold, and W.R. Cotton, 2002: Simulations of aerosol-cloud-dynamical feedbacks resulting from of entrainment of aerosols into the marine boundary layer during the Atlantic Stratocumulus Transition Experiment. J. Geophys. Res., 107(D24), 4813, doi:10.1029/2001JD001502. [JoC, MoS]
Jirikowic, J.L., and P.E. Damon, 1994: The Medieval solar activity maximum. Clim. Change, 26,309–316. [JoC]
Johnson, B.T., 2005: The semidirect aerosol effect: comparison of a single-column model with large eddy simulation for marine stratocumulus. J. Clim., 18,119–130. [JoC, MoS]
Johnson, B.T., K.P. Shine, and P.M. Forster, 2004: The semi-direct aerosol effect: Impact of absorbing aerosols on marine stratocumulus. Q. J. R. Meteorol. Soc., 130,1407–1422. [JoC]
Jones, A., D.L. Roberts, and M.J. Woodage, 2001: Indirect sulphate aerosol forcing in a climate model with an interactive sulphur cycle. J. Geophys. Res., 106(D17), 20293–20301. [JoC, MoS, ARC]
Jones, P.D., A. Moberg, T.J. Osborn, and K.R. Briffa, 2003: Surface climate responses to explosive volcanic eruptions seen in long European temperature records and mid-to-high latitude tree-ring density around the Northern Hemisphere. In: Volcanism and the Earth’s Atmosphere [Robock, A., and C. Oppenheimer (eds.)]. Geophysical Monograph 139, American Geophysical Union, Washington, DC, pp. 239–254. [NPR, PoC, ARC]
Jonson, J.E., D. Simpson, H. Fagerli, and S. Solberg, 2005: Can we explain the trends in European ozone levels? Atmos. Chem. Phys. Discuss., 5,5957–5985. Clean
Joos, F., et al., 1996. An efficient and accurate representation of complex oceanic and biospheric models of anthropogenic carbon uptake. Tellus, 48B,397–417. [PoC, JoC, MoS, SRC]
Joos, F., et al., 2001. Global warming feedbacks on terrestrial carbon uptake under the Intergovernmental Panel on Climate Change (IPCC) emission scenarios. Global Biogeochem. Cycles, 15,891-908, 2001 [PoC, JoC, SRC]
Joshi, M.M., and K.P. Shine, 2003: A GCM study of volcanic eruptions as a cause of increased stratospheric water vapor. J. Clim., 16,3525–3534. [JoC]
Joshi, M., et al., 2003: A comparison of climate response to different radiative forcings in three general circulation models: Towards an improved metric of climate change. Clim. Dyn., 20,843–854. [JoC, MoS]
Junge, C.E., 1975: The possible influence of aerosols on the general circulation and climate and possible approaches for modelling. In: The Physical Basis of Climate and Climate Modelling: Report of the International Study Conference in Stockholm, 29 July-10 August 1974: organised by WMO and ICSU and supported by UNEP Global Atmospheric Research Programme (GARP), WMO-ICSU Joint Organising Committee.World Meteorological Organization, Geneva, pp. 244–251. [NPR, MoS]
Kahn, R., P. Banerjee, and D. McDonald, 2001: Sensitivity of multiangle imaging to natural mixtures of aerosols over ocean. J. Geophys. Res., 106(D16), 18219–18238. [JoC]
Kahn, R.A., et al., 2005: Multiangle Imaging Spectroradiometer (MISR) global aerosol optical depth validation based on 2 years of coincident Aerosol Robotic Network (AERONET) observation. J. Geophys. Res., 110,D10S04, doi:10.1029/2004JD004706. [JoC]
Kalkstein, A.J., and R.C. Balling Jr., 2004: Impact of unusually clear weather on United States daily temperature range following 9/11/2001. Clim. Res., 26,1–4. Clean
Kanakidou, M., et al., 2005: Organic aerosol and global climate modelling: a review. Atmos. Chem. Phys., 5,1053–1123. [MoS]
Kandlikar, M., 1996: Indices for comparing greenhouse gas emissions: integrating science and economics. Energy Econ., 18,265–282. Clean
Kapustin, V.N., et al., 2006: On the determination of a cloud condensation nuclei from satellite: challenges and possibilities. J. Geophys. Res., 111,D04202, doi:10.1029/2004JD005527. [JoC]
Kärcher, B., 1999: Aviation-produced aerosols and contrails. Surv. Geophys., 20,113–167. [ARC]
Kasischke, E.S., and J.E. Penner, 2004: Improving global estimates of atmospheric emissions from biomass burning. J. Geophys. Res., 109,D14S01, doi:10.1029/2004JD004972. [JoC, MoS, ARC]
Kaufman, Y.J., C.J. Tucker, and R.L Mahoney, 1991: Fossil fuel and biomass burning effect on climate: Heating or cooling? J. Clim., 4,578–588. [JoC]
Kaufman, Y.J., D. Tanré, and O. Boucher, 2002: A satellite view of aerosols in the climate system. Nature, 419,215–223. [JoC, SRC]
Kaufman, Y.J., et al., 1997: Operational remote sensing of tropospheric aerosols over the land from EOS-MODIS. J. Geophys. Res., 102(D14), 17051–17068. [JoC]
Kaufman, Y.J., et al., 2001: Absorption of sunlight by dust as inferred from satellite and ground-based remote sensing. Geophys. Res. Lett., 28(8), 1479–1482. [JoC]
Kaufman Y.J., et al., 2005a: Aerosol anthropogenic component estimated from satellite data. Geophys. Res. Lett., 32,L17804, doi:10.1029/2005GL023125. [JoC, MoS]
Kaufman, Y.J., et al., 2005b: A critical examination of the residual cloud contamination and diurnal sampling effects on MODIS estimates of aerosol over ocean. IEEE Trans. Geosci. Remote, 43(12), 2886–2897. [MoS]
Keeling, C.D., and T.P. Whorf, 2005: Atmospheric CO2 records from sites in the SIO air sampling network. In: Trends: A Compendium of Data on Global Change.Carbon Dioxide Information Analysis Center, Oak Ridge National Laboratory, U.S. Department of Energy, Oak Ridge, TN, http://cdiac.esd.ornl.gov/trends/co2/sio-keel-flask/sio-keel-flask.html. [NPR, SRC]
Keeling, C.D., A.F. Bollenbacher, and T.P. Whorf, 2005: Monthly atmospheric 13C/12Cisotopic ratios for 10 SIO stations. In: Trends: A Compendium of Data on Global Change.Carbon Dioxide Information Analysis Center, Oak Ridge National Laboratory, U.S. Department of Energy, Oak Ridge, TN, http://cdiac.ornl.gov/trends/co2/iso-sio/iso-sio.html. [NPR, SRC]
Keeling, C.D., T.P. Whorf, M. Wahlen, and J. van der Plicht, 1995: Interannual extremes in the rate of rise of atmospheric carbon dioxide since 1980. Nature, 375,666–670. [JoC, SRC]
Keeling, R.F., and S.R. Shertz, 1992: Seasonal and interannual variations in atmospheric oxygen and implications for the global carbon cycle. Nature, 358,723–727. [JoC, SRC]
Keeling, R.F., S.C. Piper, and M. Heimann, 1996: Global and hemispheric CO2 sinks deduced from changes in atmosphericO2 concentration. Nature, 381,218–221. [JoC, SRC]
Keil, A., and J.M. Haywood, 2003: Solar radiative forcing by biomass burning aerosol particles during SAFARI 2000: A case study based on measured aerosol and cloud properties. J. Geophys. Res., 108 (D13), 8467, doi:10.1029/2002JD002315. [JoC, MoS, SRC]
Keppler, F., J.T.G. Hamilton, M. Brass, and T. Röckmann, 2006: Methane emissions from terrestrial plants under aerobic conditions. Nature, 439,187–191, doi:10.1038/nature04420. [JoC]
Kernthaler, S.C., R. Toumi, and J.D. Haigh, 1999: Some doubts concerning a link between cosmic ray fluxes and global cloudiness. Geophys. Res. Lett., 26(7), 863–866, doi:10.1029/1999GL900121. [JoC, ARC]
Khainroutdinov, M., and Y. Kogan, 2000: A new cloud physics parametrization in a large-eddy simulation model of marine stratocumulus. Mon. Weather Rev., 128,229–243. [JoC, MoS]
Khalil, M.A.K., et al., 2003: Atmospheric perfluorocarbons. Environ. Sci. Technol., 37,4358–4361. Clean
Kiehl, J.T., et al., 2000: Radiative forcing due to sulfate aerosols from simulations with the National Center for Atmospheric Research Community Climate Model, Version 3. J. Geophys. Res., 105(D1), 1441–1457. [JoC, MoS]
Kim, B.-G., S.E. Schwartz, and M.A. Miller, 2003: Effective radius of cloud droplets by ground-based remote sensing: Relationship to aerosol. J. Geophys. Res., 108(D23), doi:10.1029/2003JD003721. [JoC, ARC]
Kinne, S., et al., 2003: Monthly averages of aerosol properties: a global comparison among models, satellite data, and AERONET ground data. J. Geophys. Res., 108 (D20), 4634, doi:10.1029/2001JD001253. [JoC, MoS, SRC]
Kinne, S., et al., 2006: An AeroCom initial assessment: optical properties in aerosol component modules of global models. Atmos. Chem. Phys. 6,1815–1834. [MoS, SRC]
Kirchstetter, T.W., T. Novakov, and P.V. Hobbs, 2004: Evidence that the spectral dependence of light absorption by aerosols is affected by organic carbon. J. Geophys. Res., 109,D21208, doi:10.1029/2004JD004999. [JoC]
Kirkevåg, A., and T. Iversen, 2002: Global direct radiative forcing by process-parametrized aerosol optical properties. J. Geophys. Res., 107(D20), 4433, doi:10.1029/2001JD000886. [JoC, MoS]
Klein Goldewijk, K., 2001: Estimating global land use change over the past 300 years: The HYDE database. Global Biogeochem. Cycles, 15,417–433. [JoC, MoS]
Knutson, T.R., et al., 2006: Assessment of twentieth-century regional surface temperature trends using the GFDL CM2 coupled models. J. Clim., 10(9), 1624–1651. [JoC, MoS, ARC]
Koch, D., 2001: Transport and direct radiative forcing of carbonaceous and sulfate aerosols in the GISS GCM. J. Geophys. Res., 106(D17), 20311–20332. [JoC, MoS]
Korolev, A.V., and G.A. Isaac, 2005: Shattering during sampling OAPs and HVPS. Part I: snow particles. J. Atmos. Ocean. Technol., 22,528–542. Clean
Kotchenruther, R.A., and P.V. Hobbs, 1998: Humidification factors of aerosols from biomass burning in Brazil. J. Geophys. Res., 103(D24), 32081–32089. [JoC]
Kotchenruther, R.A., P.V. Hobbs, and D.A. Hegg, 1999: Humidification factors for atmospheric aerosols off the mid-Atlantic coast of the United States. J. Geophys. Res., 104(D2), 2239–2251. [JoC]
Kristjánsson, J.E., and J. Kristiansen, 2000: Is there a cosmic ray signal in recent variations in global cloudiness and cloud radiative forcing? J. Geophys. Res., 105(D9), 11851–11863. [JoC, MoS]
Kristjánsson, J.E., A. Staple, J. Kristiansen, and E. Kaas, 2002: A new look at possible connections between solar activity, clouds and climate. Geophys. Res. Lett., 29,doi:10.1029/2002GL015646. [NPR, JoC]
Kristjánsson, J.E., et al., 2005: Response of the climate system to aerosol direct and indirect forcing: the role of cloud feedbacks. J. Geophys. Res., 110,D24206, doi:10.1029/2005JD006299. [JoC, MoS]
Krizan P., and J. Lastovicka, 2005: Trends in positive and negative ozone laminae in the Northern Hemisphere. J. Geophys. Res., 110,D10107, doi:10.1029/2004JD005477. [JoC]
Kroeze, C., E. Dumont, and S.P. Seitzinger, 2005: New estimates of global emissions ofN2Ofrom rivers, estuaries and continental shelves. Environ. Sci.,2(2–3), 159–165. [MoS]
Krol, M., and J. Lelieveld, 2003: Can the variability in tropospheric OH be deduced from measurements of 1,1,1-trichloroethane (methyl chloroform)? J. Geophys. Res., 108(D3), 4125, doi:10.1029/2002JD002423. [JoC]
Krol, M., et al., 2003: Continuing emissions of methyl chloroform from Europe. Nature, 421,131–135. [JoC]
Krueger, A.J., et al., 2000: Ultraviolet remote sensing of volcanic emissions. In: Remote Sensing of Active Volcanism [Mouginis-Mark, P.J., J.A. Crisp, and J.H. Fink (eds.)]. Geophysical Monograph 116, American Geophysical Union, Washington, DC, pp. 25–43. [NPR]
Krüger, O., and H. Graßl, 2002: The indirect aerosol effect over Europe. Geophys. Res. Lett., 29,doi:10.1029/2001GL14081. [NPR, JoC]
Labitzke, K., 2004: On the signal of the 11-year sunspot cycle in the stratosphere and its modulation by the quasi-biennial oscillation. J. Atmos. Solar Terr. Phys., 66,1151–1157. Clean
Labitzke, K., et al., 2002: The global signal of the 11-year solar cycle in the stratosphere: observations and models. J. Atmos. Solar Terr. Phys., 64,203–210. [MoS]
Lambert, A., et al., 1993: Measurements of the evolution of the Mt. Pinatubo aerosol cloud by ISAMS. Geophys. Res. Lett., 20(12), 1287–1290. [JoC]
Langenfelds, R.L., et al., 2002: Interannual growth rate variations of atmospheric CO2 and its delta C-13, H-2, CH4,and CO between 1992 and 1999 linked to biomass burning. Global Biogeochem. Cycles, 16,doi:10.1029/2001GB001466. [JoC]
Lawrence, M.G., P. Jöckel, and R. von Kuhlmann, 2001: What does the global mean OH concentration tell us? Atmos. Chem. Phys., 1,43–74. Clean
Lean, J., 2000: Evolution of the sun’s spectral irradiance since the Maunder Minimum. Geophys. Res. Lett., 27,2425–2428. [JoC, SRC]
Lean, J., A. Skumanich, and O.White, 1992: Estimating the sun’s radiative output during the Maunder Minimum. Geophys. Res. Lett., 19(15), 1595–1598. [JoC, MoS, SRC]
Lean, J., J. Beer, and R. Bradley, 1995: Reconstruction of solar irradiance since 1610: implications for climate change. Geophys. Res. Lett., 22,3195–3198. [PoC, JoC, SRC]
Lean, J.L., Y.M. Wang, and N.R. Sheeley, 2002: The effect of increasing solar activity on the Sun’s total and open magnetic flux during multiple cycles: Implications for solar forcing of climate. Geophys. Res. Lett., 29(24), 2224, doi:10.1029/2002GL015880. [JoC, MoS, SRC]
Lean, J., G. Rottman, J. Harder, and G. Kopp, 2005: SORCE contribution to new understanding of global change and solar variability. Solar Phys., 230,27–53. [SRC]
Lean, J.L., et al., 1997: Detection and parametrization of variations in solar mid and near ultraviolet radiation (200 to 400 nm). J. Geophys. Res., 102(D25), 29939–29956. [JoC, SRC]
Lee, K.H., Y.J. Kim, and W. von Hoyningen-Huene, 2004: Estimation of regional aerosol optical thickness from satellite observations during the 2001 ACE-Asia IOP. J. Geophys. Res. 109, D19S16, doi:10.1029/2003JD004126. [JoC, MoS]
Lee, R.B. III, M.A. Gibson, R.S. Wilson, and S. Thomas, 1995: Long-term total solar irradiance variability during sunspot cycle 22. J. Geophys. Res., 100(A2), 1667–1675. [JoC]
Lefohn, A.S., J.D. Husar, and R.B. Husar, 1999: Estimating historical anthropogenic global sulfur emission patterns for the period 1850-1990. Atmos. Environ., 33,3435–3444. [MoS]
Lelieveld, J., P.J. Crutzen, and F.J. Dentener, 1998: Changing concentration, lifetime and climate forcing of atmospheric methane. Tellus, 50B,128–150. [JoC, MoS, ARC]
Lelieveld, J., W. Peters, F.J. Dentener, and M.C. Krol, 2002: Stability of tropospheric hydroxyl chemistry. J. Geophys. Res., 107(D23), 4715, doi:10.1029/2002JD002272. [JoC, ARC]
Lelieveld, J., et al., 2004: Increasing ozone over the Atlantic Ocean. Science, 304,1483–1487. [JoC]
Levy, R.C., et al., 2003: Evaluation of the Moderate-Resolution Imaging Spectroradiometer (MODIS) retrievals of dust aerosol over the ocean during PRIDE. J. Geophys. Res., 108(D19), 8594, doi:10.1029/2002JD002460. [JoC]
Li, J., et al., 2005: Halocarbon emissions estimated from AGAGE measured pollution events at Trinidad Head, California. J. Geophys. Res., 110,D14308, doi:10.1029/2004JD005739. [JoC, MoS]
Liao, H., and J.H. Seinfeld, 2005: Global impacts of gas-phase chemistry-aerosol interactions on direct radiative forcing by anthropogenic aerosols and ozone. J. Geophys. Res., 110,D18208, doi:10.1029/2005JD005907. [JoC, MoS]
Liao, H., J.H. Seinfeld, P.J. Adams, and L.J. Mickley, 2004: Global radiative forcing of coupled tropospheric ozone and aerosols in a unified general circulation model. J. Geophys. Res., 109,D16207, doi:10.1029/2003JD004456. [JoC, MoS]
Liao, T., C.D. Camp, and Y.L. Yung, 2004: The seasonal cycle ofN2O. Geophys. Res. Lett., 31,L17108, doi:10.1029/2004GL020345. [JoC]
Liousse, C., et al., 1996: A global three-dimensional model study of carbonaceous aerosols. J. Geophys. Res., 101(D14), 19411–19432. [JoC, MoS]
Liu, H.Q., R.T. Pinker, and B.N. Holben, 2005: A global view of aerosols from merged transport models, satellite, and ground observations. J. Geophys. Res., 110(D10), doi:10.1029/2004JD004695. [JoC, MoS]
Liu, X.H., and J.E. Penner, 2002: Effect of Mount PinatuboH2SO4/H2Oaerosol on ice nucleation in the upper troposphere using a global chemistry and transport model. J. Geophys. Res., 107(D12), doi:10.1029/2001JD000455. [JoC, MoS, ARC]
Liu, Y., and P.H. Daum, 2002: Indirect warming effect from dispersion forcing. Nature, 419,580–581. [JoC, MoS]
Lockwood, M., and R. Stamper, 1999: Long-term drift of the coronal source magnetic flux and the total solar irradiance. Geophys. Res. Lett., 26(16), 2461–2464. [JoC]
Loeb, N.G., and S. Kato, 2002: Top-of-atmosphere direct radiative effect of aerosols over the tropical oceans from the Clouds and the Earth’s Radiant Energy System (CERES) satellite instrument. J. Clim., 15,1474–1484. [JoC]
Loeb, N.G., and N. Manalo-Smith, 2005: Top-of-atmosphere direct radiative effect of aerosols over global oceans from merged CERES and MODIS observations. J. Clim., 18,3506–3526. [JoC]
Lohar, D., and B. Pal, 1995: The effect of irrigation on premonsoon season precipitation over south west Bengal, India. J. Clim., 8,2567–2570. [JoC]
Lohmann, U., and G. Lesins, 2002: Stronger constraints on the anthropogenic indirect aerosol effect. Science, 298,1012–1016. [JoC, SRC]
Lohmann, U., and J. Feichter, 2005: Global indirect aerosol effects: A review. Atmos. Chem. Phys., 5,715–737. [SRC]
Lohmann, U. and C. Leck, 2005: Importance of submicron surface active organic aerosols for pristine Arctic clouds. Tellus, 57B,261–268. [JoC, SRC]
Lohmann, U., B. Kärcher, and J. Hendrichs, 2004: Sensitivity studies of cirrus clouds formed by heterogeneous freezing in ECHAM GCM. J. Geophys. Res., 109,D16204, doi:10.1029/2003JD004443. [JoC, SRC]
Lohmann, U., J. Feichter, J.E. Penner, and W.R. Leaitch, 2000: Indirect effect of sulfate and carbonaceous aerosols: a mechanistic treatment. J. Geophys. Res., 105(D10), 12193–12206. [JoC, SRC]
Lohmann, U., et al., 2001: Vertical distributions of sulfur species simulated by large scale atmospheric models in COSAM: comparison with observations. Tellus, 53B,646–672. [JoC, MoS, SRC]
Loveland, T.R., et al., 2000: Development of a global land cover characteristics database and IGBP DISCover from 1 km AVHR R data. Int. J. Remote Sens., 21,1303–1330. Clean
Lowe, D.C., and W. Allan, 2002: A simple procedure for evaluating global cosmogenic 14Cproduction in the atmosphere using neutron monitor data. Radiocarbon, 44,149–157. [SRC]
Lowe, D.C., M.R. Manning, G.W. Brailsford, and A.M. Bromley, 1997: The 1991-1992 atmospheric methane anomaly: Southern hemisphere 13Cdecrease and growth rate fluctuations. Geophys. Res. Lett., 24(8), 857–860. [JoC, SRC]
Lowe, D.C., et al., 2004: Seasonal cycles of mixing ratio and 13Cin atmospheric methane at Suva, Fiji. J. Geophys. Res., 109,D23308 doi:10.1029/2004JD005166. [JoC, SRC]
Lu, M.-L., and J. Seinfeld, 2005: Study of the aerosol indirect effect by large-eddy simulation of marine stratocumulus. J. Atmos. Sci., 62,3909–3932. [JoC, MoS]
Luo, C., N.M. Mahowald, and J. del Corral, 2003: Sensitivity study of meteorological parameters on mineral aerosol mobilization, transport, and distribution, J. Geophys. Res., 108 (D15), 4447, doi:10.1029/2003JD003483. [JoC]
MacFarling Meure, C., et al., 2006: The Law Dome CO2,CH4 andN2Oice core records extended to 2000 years BP. Geophys. Res. Lett., 33,L14810, doi:10.1029/2006GL026152. [JoC]
Macke, A., M. Mishchenko, and B. Cairns, 1996: The influence of inclusions on light scattering by large ice particles. J. Geophys. Res., 101(D18), 23311–23316. [JoC]
Magi, B.I., and P.V. Hobbs, 2003: Effects of humidity on aerosols in southern Africa during the biomass burning season. J. Geophys. Res., 108(D13), 8495, doi:10.1029/2002JD002144. [JoC]
Magi, B.I., et al., 2005: Properties and chemical apportionment of aerosol optical depth at locations off the U.S. East Coast in July and August 2001. J. Atmos. Sci., 62(4), 919–933, doi:10.1175/JAS3263.1. [JoC]
Mahowald, N.M., and C. Luo, 2003: A less dusty future? Geophys. Res. Lett., 30(17), doi:10.1029/2003GL017880. [JoC, MoS]
Mahowald, N.M., G.C. Rivera, and C. Luo, 2004: Comment on Tegen et al. 2004, on the “Relative importance of climate and land use in determining present and future global soil dust emissions”. Geophys. Res. Lett., 31,L24105, doi:10.1029/2004GL021272. [JoC, MoS]
Mak, J.E., M.R. Manning, and D.C. Lowe, 2000: Aircraft observations of δ13Cof atmospheric methane over the Pacific in August 1991 and 1993: evidence of an enrichment in 13CH4 in the Southern Hemisphere. J. Geophys. Res., 105(D1), 1329–1335. [JoC, SRC]
Malm, W.C., et al., 2004: Spatial and monthly trends in speciated fine particle concentration in the United States. J. Geophys. Res., 109,D03306, doi:10.1029/2003JD003739. [JoC]
Manabe, S., and R.T. Wetherald, 1967: Thermal equilibrium of the atmosphere with a given distribution of relative humidity. J. Atmos. Sci., 24,241–259. [JoC]
Manne, A.S., and R.G. Richels, 2001: An alternative approach to establishing trade-offs among greenhouse gases. Nature, 410,675–676. [JoC]
Manning, A.C., and R.F. Keeling, 2006: Global oceanic and land biotic carbon sinks from the Scripps atmospheric oxygen flask sampling network. Tellus, 58B,95–116. [JoC, SRC]
Manning, A.J., et al., 2003: Estimating European emissions of ozone-depleting and greenhouse gases using observations and a modelling back-attribution technique. J. Geophys. Res., 108 (D14), 4405, doi:10.1029/2002JD002312. [JoC, MoS]
Manning, M.R., A. Gomez, and G.W. Brailsford, 1997: Annex B11: The New Zealand CO2 measurement programme. In: Report of the Ninth WMO Meeting of Experts on Carbon Dioxide Concentration and Related Tracer Measurement Techniques. WMO Global Atmosphere Watch No. 132; WMO TD No. 952, Commonwealth Scientific and Industrial Research Organisation, Melbourne, pp. 120–123. [NPR, ARC]
Manning, M.R., et al., 2005: Short term variations in the oxidizing power of the atmosphere. Nature, 436,1001–1004. [JoC, ARC]
Mannstein, H., and U. Schumann, 2005: Aircraft induced contrail cirrus over Europe. Meteorol. Z., 14,549–544. Clean
Maria, S., F. Russell, L.M. Turpin, and R.J. Porcja, 2002: FTIR measurements of functional groups and organic mass in aerosol samples over the Caribbean. Atmos. Environ., 36,5185–5196. Clean
Marland, G., T.A. Boden, and R.J. Andres, 2006: Global, regional, and national CO2 emissions. In: Trends: A Compendium of Data on Global Change.Carbon Dioxide Information Analysis Center, Oak Ridge National Laboratory, U.S. Department of Energy, Oak Ridge, TN, http://cdiac.esd.ornl.gov/trends/emis/tre_glob.htm. [NPR]
Marquart, S., and B. Mayer, 2002: Towards a reliable GCM estimation of contrail radiative forcing. Geophys. Res. Lett., 29(8), doi:10.1029/2001GL014075. [JoC, MoS]
Marquart, S., M. Ponater, F. Mager, and R. Sausen, 2003: Future development of contrail cover, optical depth, and radiative forcing: Impacts of increasing air traffic and climate change. J. Clim., 16,2890–2904. [JoC, MoS, SRC]
Marsh, N.D., and H. Svensmark, 2000a: Low cloud properties influenced by cosmic rays. Phys. Rev. Lett., 85,5004–5007. Clean
Marsh, N.D., and H. Svensmark, 2000b: Cosmic rays, clouds, and climate. Space Sci. Rev., 94,215–230. Clean
Marshak, A., et al., 2006: Impact of three-dimensional radiative effects on satellite retrievals of cloud droplet sizes. J. Geophys. Res., 111,D09207, doi:10.1029/2005JD006686. [JoC]
Martin, G.M., D.W. Johnson, and A. Spice, 1994: The measurement and parametrization of effective radius of droplets in warm stratiform clouds. J. Atmos. Sci., 51,1823–1842. [JoC]
Martin, S.T., et al., 2004: Effects of the physical state of tropospheric ammonium-sulfate-nitrate particles on global aerosol direct radiative forcing. Atmos. Chem. Phys., 4,183–214. [MoS]
Martonchik, J.V., et al., 2004: Comparison of MISR and AERONET aerosol optical depths over desert sites. Geophys. Res. Lett., 31,L16102, doi:10.1029/2004GL019807. [JoC]
Matthews, E., 1983: Global vegetation and land-use: new high-resolution data-bases for climate studies. J. Clim. Appl. Meteorol., 22,474–487. Clean
Matthews, H.D., A.J. Weaver, M. Eby, and K.J. Meissner, 2003: Radiative forcing of climate by historical land cover change. Geophys. Res. Lett., 30(2), 271–274. [JoC, MoS, ARC]
Matthews, H.D., et al., 2004: Natural and anthropogenic climate change: Incorporating historical land cover change, vegetation dynamics and the global carbon cycle. Clim. Dyn., 22,461–479. [JoC, ARC]
Matthias, I., et al., 2004: Multiyear aerosol observations with dual wavelength Raman lidar in the framework of EARLINET. J. Geophys. Res., 109,D13203, doi:10.1029/2004JD004600. [JoC]
McCormack, J.P., 2003: The influence of the 11-year solar cycle on the quasi-biennial oscillation. Geophys. Res. Lett., 30 (22), 2162, doi:10.1029/2003GL018314. [JoC]
McCormick, M.P., 1987: SAGE II: An overview. Adv. Space Res., 7,219–226. Clean
McCulloch, A., and P.M. Midgley, 2001: The history of methyl chloroform emissions: 1951-2000. Atmos. Environ., 35,5311–5319. Clean
McFiggans, G., et al., 2006: The effect of aerosol composition and properties on warm cloud droplet activation. Atmos. Chem. Phys., 6,2593–2649. [ARC]
Meehl, G.A., et al., 2004: Combinations of natural and anthropogenic forcings in twentieth-century climate. J. Clim., 17,3721–3727. [JoC, MoS, ARC]
Meerkötter, R., et al., 1999: Radiative forcing by contrails. Ann. Geophys., 17,1080–1094. [MoS]
Melillo, J.M., et al., 2001: Nitrous oxide emissions from forests and pastures of various ages in the Brazilian Amazon. J. Geophys. Res., 106(D24), 34179–34188. [JoC]
Menon, S., and A. Del Genio, 2007: Evaluating the impacts of carbonaceous aerosols on clouds and climate. In: Human-Induced Climate Change: An Interdisciplinary Assessment [Schlesinger, M., et al. (eds.)]. Cambridge University Press, Cambridge, UK, in press. [NPR, ARC, 2007]
Menon, S., A.D. Del Genio, D. Koch, and G. Tselioudis, 2002a: GCM simulations of the aerosol indirect effect: sensitivity to cloud parametrization and aerosol burden. J. Atmos. Sci., 59,692–713. [JoC, MoS, ARC]
Menon, S., J. Hansen, L. Nazarenko, and Y. Luo, 2002b: Climate effects of black carbon aerosols in China and India. Science, 297,2250–2253. [JoC, ARC]
Menon, S., et al., 2003: Evaluating aerosol/cloud/radiation process parametrizations with single-column models and Second Aerosol Characterization Experiment (ACE-2) cloudy column observations. J. Geophys. Res., 108(D24), 4762, doi:10.1029/2003JD003902. [JoC, MoS, ARC]
Meyer, R., et al., 2002: Regional radiative forcing by line-shaped contrails derived from satellite data. J. Geophys. Res., 107(D10), 4104, doi:10.1029/2001JD000426. [JoC, MoS]
Mickley, L.J., D.J. Jacob, and D. Rind, 2001: Uncertainty in preindustrial abundance of tropospheric ozone: implications for radiative forcing calculations. J. Geophys. Res., 106(D4), 3389–3399 doi:10.1029/2000JD900594. [JoC, MoS, SRC]
Mickley, L.J., D.J. Jacob, B.D. Field, and D. Rind, 2004: Climate response to the increase in tropospheric ozone since preindustrial times: a comparison between ozone and equivalent CO2 forcings. J. Geophys. Res., 109,D05106, doi:10.1029/2003JD003653. [JoC, MoS, SRC]
Mikami, M., et al., 2006: Aeolian dust experiment on climate impact: an overview of Japan-China Joint Project ADEC. Global Planet. Change, 52,142–172, doi:10.1016/j.gloplacha.2006.03.001. [JoC]
Miller, B.R., et al., 1998: Atmospheric trend and lifetime of chlorodifluoromethane (HCFC-22) and the global tropospheric OH concentration. J. Geophys. Res., 103(D11), 13237–13248, doi:10.1029/98JD00771. [JoC]
Miller, R.L., G.A. Schmidt, and D.T. Shindell, 2006: Forced annular variations in the 20th century IPCC AR4 models. J. Geophys. Res., 111,D18101, doi:10.1029/2005JD006323. [PoC, JoC, MoS, ARC]
Millet, D.B., and A.H. Goldstein, 2004: Evidence of continuing methylchloroform emissions from the United States. Geophys. Res. Lett., 31,L17101, doi:10.1029/2004GL020166. [JoC]
Ming, Y., and L.M. Russell, 2002: Thermodynamic equilibrium of organic-electrolyte mixtures in aerosol particles. Am. Inst. Chem. Eng. J., 48,1331–1348. Clean
Ming, Y., V. Ramaswamy, P.A. Ginoux, and L.H. Horowitz, 2005a: Direct radiative forcing of anthropogenic organic aerosol. J. Geophys. Res., 110,D20208, doi:10.1029/2004JD005573. [JoC, MoS, SRC]
Ming, Y., V. Ramaswamy, L.J. Donner, and V.T.J. Phillips, 2006: A new parametrization of cloud droplet activation applicable to general circulation models. J. Atmos. Sci., 63(4), 1348–1356. [JoC, MoS, SRC]
Ming, Y., et al., 2005b: Geophysical Fluid Dynamics Laboratory general circulation model investigation of the indirect radiative effects of anthropogenic sulfate aerosol. J. Geophys. Res., 110,D22206, doi:10.1029/2005JD006161. [JoC, MoS]
Ming, Y., et al., 2007: Modelling the interactions between aerosols and liquid water clouds with a self-consistent cloud scheme in a general circulation model. J. Atmos. Sci., 64(4), 1189–1209. [JoC, MoS, 2007]
Minnis, P., 2005: Reply. J. Clim., 18,2783–2784. [JoC]
Minnis, P., J.K. Ayers, R. Palikonda, and D. Phan, 2004: Contrails, cirrus trends, and climate. J. Clim., 17,1671–1685. [JoC]
Minnis, P., et al., 1993: Radiative climate forcing by the Mt. Pinatubo eruption. Science, 259,1411–1415. [JoC, MoS]
Minnis, P., et al., 1998: Transformation of contrails into cirrus during SUCCESS. Geophys. Res. Lett., 25,1157–1160. [JoC]
Mircea, M., et al., 2005: Importance of the organic aerosol fraction for modeling aerosol hygroscopic growth and activation: a case study in the Amazon Basin. Atmos. Chem. Phys., 5,3111–3126. [MoS]
Mishchenko, M.I., et al., 1999: Aerosol retrievals over the ocean by use of channels 1 and 2 AVHRR data: sensitivity analysis and preliminary results. Appl. Opt., 38,7325–7341. Clean
Möhler, O., et al., 2005: Effect of sulfuric acid coating on heterogeneous ice nucleation by soot aerosol particles. J. Geophys. Res., 110,D11210, doi:10.1029/2004JD005169. [JoC]
Montzka, S.A., et al., 1999: Present and future trends in the atmospheric burden of ozone-depleting halogens. Nature, 398,690–694. [JoC, MoS, SRC]
Montzka, S.A., et al., 2003: Controlled substances and other source gases. In: Scientific Assessment of Ozone Depletion: 2002.World Meteorological Organization, Geneva, pp. 1.1–1.83. [NPR, SRC]
Moore, N., and S. Rojstaczer, 2001: Irrigation-induced rainfall and the Great Plains. J. Appl. Meteorol., 40,1297–1309. [JoC]
Morgan, C.G., et al., 2004: Isotopic fractionation of nitrous oxide in the stratosphere: Comparison between model and observations. J. Geophys. Res., 109,D04305, doi:10.1029/2003JD003402. [JoC, MoS]
Morimoto, S., S. Aoki, T. Nakazawa, and T. Yamanouchi , 2006: Temporal variations of the carbon isotopic ratio of atmospheric methane observed at Ny Ålesund, Svalbard from 1996 to 2004. Geophys. Res. Lett., 33,L01807, doi:10.1029/2005GL024648. [JoC]
Mosley-Thompson, E., T.A. Mashiotta, and L.G. Thompson, 2003: High resolution ice core records of Late Holocene volcanism: Current and future contributions from the Greenland PARCA cores. In: Volcanism and the Earth’s Atmosphere [Robock, A., and C. Oppenheimer (eds.)]. Geophysical Monograph 139, American Geophysical Union, Washington, DC, pp. 153–164. [NPR, MoS, ARC]
Mouillot, F., et al., 2006: Global carbon emissions from biomass burning in the 20th century. Geophys. Res. Lett., 33,L01801, doi:10.1029/2005GL024707. [JoC]
Moulin, C., and I. Chiapello, 2004: Evidence of the control of summer atmospheric transport of African dust over the Atlantic by Sahel sources from TOMS satellites (1979-2000). Geophys. Res. Lett., 31,L02107, doi:10.1029/2003GL018931. [JoC]
Moulin, C., H.R. Gordon, V.F. Banzon, and R.H. Evans, 2001: Assessment of Saharan dust absorption in the visible from SeaWiFS imagery. J. Geophys. Res., 106(D16), 18239–18249. [JoC]
Murayama, T., et al., 2001: Ground-based network observation of Asian dust events of April 1998 in East Asia. J. Geophys. Res., 106,18345–18360. [JoC]
Murphy, D.M., 2005: Something in the air. Science, 307,1888–1890. [JoC]
Muscheler, R., et al., 2007: Solar activity during the last 1000 yr inferred from radionuclide records. Quat. Sci. Rev., 26,82-97, doi:10.1016/j.quascirev.2006.07.012. [JoC, ARC, 2007]
Myhre, G., and F. Stordal, 2001a: Global sensitivity experiments of the radiative forcing due to mineral aerosols. J. Geophys. Res., 106,18193–18204. [JoC, MoS, SRC]
Myhre, G., and F. Stordal, 2001b: On the tradeoff of the solar and thermal infrared radiative impact of contrails. Geophys. Res. Lett., 28,3119–3122. [JoC, SRC]
Myhre, G., and A. Myhre, 2003: Uncertainties in radiative forcing due to surface albedo changes caused by land-use changes. J. Clim., 16,1511–1524. [JoC, MoS, SRC]
Myhre, G., A. Myhre, and F. Stordal, 2001: Historical evolution of radiative forcing of climate. Atmos. Environ., 35,2361–2373. [MoS, SRC]
Myhre, G., M.M. Kvalevåg, and C.B. Schaaf, 2005a: Radiative forcing due to anthropogenic vegetation change based on MODIS surface albedo data. Geophys. Res. Lett., 32,L21410, doi:10.1029/2005GL024004. [JoC, MoS, SRC]
Myhre, G., et al., 2003: Modelling the solar radiative impact of aerosols from biomass burning during the Southern African Regional Science Initiative (SAFARI 2000) experiment. J. Geophys. Res., 108,8501, doi:10.1029/2002JD002313. [JoC, MoS, SRC]
Myhre, G., et al., 2004a: Intercomparison of satellite retrieved aerosol optical depth over ocean. J. Atmos. Sci., 61,499–513. [JoC, SRC]
Myhre, G., et al., 2004b: Uncertainties in the radiative forcing due to sulfate aerosols. J. Atmos. Sci., 61,485–498. [JoC, MoS, SRC]
Myhre, G., et al., 2005b: Intercomparison of satellite retrieved aerosol optical depth over ocean during the period September 1997 to December 2000. Atmos. Chem. Phys., 5,1697–1719. [SRC]
Naja, M., and H. Akimoto, 2004: Contribution of regional pollution and long-range transport to the Asia-Pacific region: Analysis of long-term ozonesonde data over Japan. J. Geophys. Res., 109,1306, doi:10.1029/2004JD004687. [JoC]
Naja, M., H. Akimoto, and J. Staehelin, 2003: Ozone in background and photochemically aged air over central Europe: analysis of long-term ozonesonde data from Hohenpeissenberg and Payerne. J. Geophys. Res., 108,4063, doi:10.1029/2002JD002477. [JoC]
Nakajima, T., and A. Higurashi, 1998: A use of two-channel radiances for an aerosol characterization from space. Geophys. Res. Lett., 25,3815–3818. [JoC, SRC]
Nakajima, T., A. Higurashi, K. Kawamoto, and J. Penner, 2001: A possible correlation between satellite-derived cloud and aerosol microphysical parameters. Geophys. Res. Lett., 28,1171–1174. [JoC, SRC]
Nakajima, T., et al., 1996: Aerosol optical properties in the Iranian region obtained by ground-based solar radiation measurements in the summer of 1991. J. Appl. Meteorol., 35,1265–1278. [JoC, SRC]
Nakazawa, T., S. Moromoto, S. Aoki, and M. Tanaka, 1997: Temporal and spatial variations of the carbon isotopic ratio of atmospheric carbon dioxide in the Western Pacific region. J. Geophys. Res., 102,1271–1285. [JoC]
Nakicenovic, N., A. Grübler, and A. McDonald (eds), 1998: Global Energy Perspectives.Cambridge University Press, New York, NY, 299 pp. [NPR]
Nenes, A., and J.H. Seinfeld, 2003: Parametrization of cloud droplet formation in global climate models. J. Geophys. Res., 108,doi:10.1029/2002JD002911. [NPR, JoC, MoS]
Nenes, A., et al., 2002: Can chemical effects on cloud droplet number rival the first indirect effect? Geophys. Res. Lett., 29,1848, doi:10.1029/2002GL015295. [JoC]
Nevison, C.D., S. Solomon, and R.S. Gao, 1999: Buffering interactions in the modeled response of stratosphericO3 to increased NOx and HOx. J. Geophys. Res., 104(D3), 3741–3754, 10.1029/1998JD100018. [PoC, JoC, MoS, ARC]
Nevison, C.D., D.E. Kinnison, and R.F. Weiss, 2004a: Stratospheric influences on the tropospheric seasonal cycles of nitrous oxide and chlorofluorocarbons. Geophys. Res. Lett., 31,L20103, doi:10.1029/2004GL020398. [JoC, SRC]
Nevison, C., T. Lueker, and R.F. Weiss, 2004b: Quantifying the nitrous oxide source from coastal upwelling. Global Biogeochem. Cycles, 18,GB1018, doi:10.1029/2003GB002110. [JoC, SRC]
Nevison, C.D., et al., 2005: Southern Ocean ventilation inferred from seasonal cycles of atmosphericN2OandO2/N2 at Cape Grim, Tasmania. Tellus, 57B,218–229. [JoC]
Newchurch, M.J., et al., 2003: Evidence for slowdown in stratospheric ozone loss: first stage of ozone recovery. J. Geophys. Res., 108(D16), 4507, doi:10.1029/2003JD003471. [JoC]
Nordhaus, W.D., 1997: Discounting in economics and climate change: an editorial comment. Clim. Change, 37,315–328. [JoC]
Notholt, J., et al., 2005: Influence of tropospheric SO2 emissions on particle formation and the stratospheric humidity. Geophys. Res. Lett., 32,L07810, doi:10.1029/2004GL022159. [JoC]
Novakov, T., D.A. Hegg, and P.V. Hobbs, 1997: Airborne measurements of carbonaceous aerosols on the East coast of the United States. J. Geophys. Res., 102(D25), 30023–20030. [JoC]
Novakov, T., et al., 2003: Large historical changes of fossil-fuel black carbon aerosols. Geophys. Res. Lett., 30(6), 1324, doi:10.1029/2002GL016345. [JoC]
Nozawa, T., T. Nagashima, H. Shiogama, and S.A. Crooks, 2005: Detecting natural influence on surface air temperature change in the early twentieth century. Geophys. Res. Lett., 32,L20719, doi:10.1029/2005GL023540. [JoC, ARC]
O’Doherty, S., et al., 2004: Rapid growth of hydrofluorocarbon 134a and hydrochlorofluorocarbons 141b, 142b, and 22 from Advanced Global Atmospheric Gases Experiment (AGAGE) observations at Cape Grim, Tasmania, and Mace Head, Ireland. J. Geophys. Res., 109,D06310, doi:10.1029/2003JD004277. [JoC]
Oinas, V., et al., 2001: Radiative cooling by stratospheric water vapor: big differences in GCM results. Geophys. Res. Lett., 28,2791–2794. [JoC]
Olivier, J.G.J., and J.J.M. Berdowski, 2001: Global emissions sources and sinks. In: The Climate System [Berdowski, J., R. Guicherit, and B.J. Heij (eds.)]. A.A. Balkema/Swets & Zeitlinger, Lisse, The Netherlands, pp. 33–78, updated at http://www.mnp.nl/edgar/. [NPR]
Oltmans, S.J., et al., 2006: Long-term changes in tropospheric ozone. Atmos. Environ., 40,3156–3173. Clean
O’Neill, B.C., 2000: The jury is still out on global warming potentials. Clim. Change, 44,427–443. [JoC]
O’Neill, B., 2003: Economics, natural science, and the costs of global warming potentials. Clim. Change, 58,251–260. [JoC]
Oram, D.E., 1999: Trends of Long-Lived Anthropogenic Halocarbons in the Southern Hemisphere and Model Calculations of Global Emissions.PhD Thesis, University of East Anglia, Norwich, UK, 249 pp. [NPR, MoS, SRC]
Oram, D.E., et al., 1998: Growth of fluoroform (CHF3,HFC-23) in the background atmosphere. Geophys. Res. Lett., 25,35–38, doi:10.1029/97GL03483. [JoC, SRC]
Osborne, S.R., J.M. Haywood, P.N. Francis, and O. Dubovik, 2004: Short-wave radiative effects of biomass burning aerosol during SAFARI2000. Q. J. R. Meteorol. Soc., 130,1423–1448. [JoC, SRC]
Palikonda, R., P. Minnis, D.P. Duda, and H. Mannstein, 2005: Contrail coverage derived from 2001 AVHRR data over the continental United States of America and surrounding area. Meteorol. Z., 14,525–536. Clean
Palmer, A.S., et al., 2002: Antarctic volcanic flux ratios from Law Dome ice cores. Ann. Glaciol., 35,329–332. Clean
Palmer, P.I., et al., 2003: Eastern Asian emissions of anthropogenic halocarbons deduced from aircraft concentration data. J. Geophys. Res., 108(D24), 4753, doi:10.1029/2003JD003591. [JoC]
Pan, W., M.A. Tatang, G.J. McRae, and R.G. Prinn, 1997: Uncertainty analysis of direct radiative forcing by anthropogenic sulfate aerosols. J. Geophys. Res., 102(D18), 21915–21924. [JoC, MoS, SRC]
Parrish, D.D., et al., 2004: Changes in the photochemical environment of the temperate North Pacific troposphere in response to increased Asian emissions. J. Geophys. Res., 109,D23S18, doi:10.1029/2004JD004978. [JoC]
Pavelin, E.G., C.E. Johnson, S. Rughooputh, and R. Toumi, 1999: Evaluation of pre-industrial surface ozone measurements made using Schönbein’s method. Atmos. Environ., 33,919–929. Clean
Peng, Y., and U. Lohmann, 2003: Sensitivity study of the spectral dispersion of the cloud droplet size distribution on the indirect aerosol effect. Geophys. Res. Lett., 30(10), 1507, doi:10.1029/2003GL017192. [JoC, SRC]
Penner, J.E., S.Y. Zhang, and C.C. Chuang, 2003: Soot and smoke aerosol may not warm climate. J. Geophys. Res., 108(D21), 4657, doi:10.1029/2003JD003409. [JoC, ARC]
Penner, J.E., X. Dong, and Y. Chen, 2004: Observational evidence of a change in radiative forcing due to the indirect aerosol effect. Nature, 427,231–234. [JoC, MoS, ARC]
Penner, J.E., et al., 2001: Aerosols, their direct and indirect effects. In: Climate Change 2001: The Scientific Basis. Contribution of Working Group I to the Third Assessment Report of the Intergovernmental Panel on Climate Change [Houghton, J.T., et al. (eds.)]. Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, pp. 289–348. [NPR, ARC]
Penner, J. E., et al., 2002: A comparison of model- and satellite-derived aerosol optical depth and reflectivity. J. Atmos. Sci., 59,441–460. [JoC, MoS, ARC]
Penner, J.E., et al., 2006: Model intercomparison of indirect aerosol effects. Atmos. Chem. Phys. Discuss., 6,1579–1617. [MoS, ARC]
Penner, J.E., et al., 2007: Effect of black carbon on mid-troposphere and surface temperature trends. In: Human-Induced Climate Change: An Interdisciplinary Assessment [Schlesinger, M., et al., (eds.)]. Cambridge University Press, Cambridge, UK, in press. [NPR, ARC, 2007]
Perlwitz, J., and H.-F. Graf, 2001: Troposphere-stratosphere dynamic coupling under strong and weak polar vortex conditions. Geophys. Res. Lett., 28,271–274. [JoC]
Perlwitz, J., and N. Harnik, 2003: Observational evidence of a stratospheric influence on the troposphere by planetary wave reflection. J. Clim., 16,3011–3026. [JoC]
Pham, M., O. Boucher, and D. Hauglustaine, 2005: Changes in atmospheric sulfur burdens and concentrations and resulting radiative forcings under IPCC SRES emission scenarios for 1990-2100. J. Geophys. Res., 110,D06112, doi:10.1029/2004JD005125. [JoC, MoS, SRC]
Philipona, R., et al., 2004: Radiative forcing - measured at Earth’s surface - corroborate the increasing greenhouse effect. Geophys. Res. Lett., 31,L03202, doi:10.1029/2003GL018765. [JoC, MoS]
Pielke, R.A. Sr., et al., 2002: The influence of land-use change and landscape dynamics on the climate system - relevance to climate change policy beyond the radiative effect of greenhouse gases. Philos. Trans. R. Soc. London Ser.A, 360,1705–1719. [JoC]
Pitari, G., E. Mancini, V. Rizi, and D.T. Shindell, 2002: Impact of future climate and emissions changes on stratospheric aerosols and ozone. J. Atmos. Sci., 59,414–440. [JoC, MoS]
Platt, U., W. Allan, and D.C. Lowe, 2004: Hemispheric average Cl atom concentration from 13C/12Cratios in atmospheric methane. Atmos. Chem. Phys., 4,2393–2399. [SRC]
Ponater, M., S. Marquart, and R. Sausen, 2002: Contrails in a comprehensive global climate model: Parametrization and radiative forcing results. J. Geophys. Res., 107(D13), doi:10.1029/2001JD000429. [JoC, MoS, SRC]
Ponater, M., S. Marquart, R. Sausen, and U. Schumann, 2005: On contrail climate sensitivity. Geophys. Res. Lett., 32,L10706, doi:10.1029/2005GL022580. [JoC, SRC]
Posfai, M., et al., 2003: Individual aerosol particles from biomass burning in southern Africa: 1. Compositions and size distributions of carbonaceous particles. J. Geophys. Res., 108(D13), 8483, doi:10.1029/2002JD002291. [JoC]
Prata, A., W. Rose, S. Self, and D. O’Brien, 2003: Global, long-term sulphur dioxide measurements from TOVS data: a new tool for studying explosive volcanism and climate. In: Volcanism and the Earth’s Atmosphere [Robock, A., and C. Oppenheimer (eds).]. Geophysical Monograph 139, American Geophysical Union, Washington, DC, pp. 75–92. [NPR]
Prather, M.J., 2003: Atmospheric science: an environmental experiment withH2? Science, 302,581–583. [JoC, ARC]
Prather, M.J., et al., 2001: Atmospheric chemistry and greenhouse gases. In: Climate Change 2001: The Scientific Basis. Contribution of Working Group I to the Third Assessment Report of the Intergovernmental Panel on Climate Change [Houghton, J.T., et al. (eds.)]. Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, pp. 239–287. [NPR, ARC]
Preminger, D.G., and S.R. Walton, 2005: A new model of total solar irradiance based on sunspot areas. Geophys. Res. Lett., 32,L14109, doi:10.1029/2005GL022839. [JoC, MoS]
Prentice, I.C., et al., 2001: The carbon cycle and atmospheric carbon dioxide. In: Climate Change 2001: The Scientific Basis. Contribution of Working Group I to the Third Assessment Report of the Intergovernmental Panel on Climate Change [Houghton, J.T., et al. (eds.)]. Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, pp. 184–238. [NPR]
Prinn, R.G., 2004: Non-CO2 greenhouse gases. In: The Global Carbon Cycle [Field, C., and M. Raupach (eds.)]. Island Press, Washington, DC, pp. 205–216. [NPR, SRC]
Prinn, R.G., et al., 1990: Atmospheric emissions and trends of nitrous oxide deduced from ten years of ALE-GAGE data. J. Geophys. Res., 95,18369–18385. [JoC, SRC]
Prinn, R.G., et al., 2000: A history of chemically and radiatively important gases in air deduced from ALE/GAGE/AGAGE. J. Geophys. Res., 105(D14), 17751–17792. [JoC, SRC]
Prinn, R.G., et al., 2001: Evidence for substantial variations of atmospheric hydroxyl radicals in the past two decades. Science, 292,1882–1888. [JoC, SRC]
Prinn, R.G., et al., 2005a: Evidence for variability of atmospheric hydroxyl radicals over the past quarter century. Geophys. Res. Lett., 32,L07809, doi:10.1029/2004GL022228. [JoC, SRC]
Prinn, R.G., et al., 2005b: The ALE/GAGE/AGAGE Network: DB1001.Carbon Dioxide Information and Analysis World Data Center, http://cdiac.esd.ornl.gov/ndps/alegage.html. [NPR, SRC]
Prospero, J.M., et al., 2002: Environmental characterization of global sources of atmospheric soil dust identified with the Nimbus 7 Total Ozone Mapping Spectrometer (TOMS) absorbing aerosol product. Rev. Geophys., 40(1), doi:10.1029/2000RG000095. [JoC]
Putaud, J.P., et al., 2004: European aerosol phenomenology-2: chemical characteristics of particulate matter at kerbside, urban, rural and background sites in Europe. Atmos. Environ., 38,2579–2595. Clean
Pyle, J., et al., 2005: Ozone and climate: a review of interconnections. In: Special Report on Safeguarding the Ozone Layer and Global Climate System: Issues Related to Hydrofluorocarbons and Perfluorocarbons [Metz., B., et al. (eds.)]. Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, pp. 83–132. [NPR]
Quaas, J., and O. Boucher, 2005: Constraining the first aerosol indirect radiative forcing in the LMDZ GCM using POLDER and MODIS satellite data. Geophys. Res. Lett., 32,L17814, doi:10.1029/2005GL023850. [JoC, MoS, SRC]
Quaas, J., O. Boucher, and F.-M. Breon, 2004: Aerosol indirect effects in POLDER satellite data and the Laboratoire de Météorologie Dynamique-Zoom (LMDZ) general circulation model. J. Geophys. Res., 109,D08205, doi:10.1029/2003JD004317. [JoC, MoS, SRC]
Quaas, J., O. Boucher, and U. Lohmann, 2005: Constraining the total aerosol indirect effect in the LMDZ and ECHAM4 GCMs using MODIS satellite data. Atmos. Chem. Phys., 5,9669–9690. [SRC]
Quay, P., et al., 2000: Atmospheric (CO)-C-14: A tracer of OH concentration and mixing rates. J. Geophys. Res., 105(D12), 15147–15166. [JoC]
Quinn, P.K., and T.S. Bates, 2005: Regional aerosol properties: comparisons from ACE 1, ACE 2, Aerosols99, INDOEX, ACE Asia, TARFOX, and NEAQS. J. Geophys. Res., 110,D14202, doi:10.1029/2004JD004755. [JoC]
Raes, F., T. Bates, F. McGovern, and M. Van Liedekerke, 2000: The 2nd Aerosol Characterization Experiment (ACE-2): general overview and main results. Tellus, 52B,111–125. [JoC]
Ramachandran, S., V. Ramaswamy, G.L. Stenchikov, and A. Robock, 2000: Radiative impact of the Mt. Pinatubo volcanic eruption: Lower stratospheric response. J. Geophys. Res., 105(D19), 24409–24429. [JoC, SRC]
Ramanathan, V., 1981: The role of ocean-atmosphere interactions in the CO2 climate problem. J. Atmos. Sci., 38,918–930. [JoC, SRC]
Ramanathan, V., P.J. Crutzen, J.T. Kiehl, and D. Rosenfeld, 2001a: Atmosphere: aerosols, climate, and the hydrological cycle. Science, 294,2119–2124. [JoC, SRC]
Ramanathan, V., et al., 2001b: Indian Ocean experiment: An integrated analysis of the climate forcing and effects of the great Indo-Asian haze. J. Geophys. Res., 106(D22), 28371–28398. [JoC, MoS, SRC]
Ramankutty, N., and J.A. Foley, 1999: Estimating historical changes in global land cover: croplands from 1700 to 1992. Global Biogeochem. Cycles, 14,997–1027. [JoC, MoS, SRC]
Ramaswamy, V., S. Ramachandran, G. Stenchikov, and A. Robock, 2006a: A model study of the effect of Pinatubo volcanic aerosols on the stratospheric temperatures. In: Frontiers of Climate Modeling [Kiehl, J.T., and V. Ramanathan (eds.)]. Cambridge University Press, Cambridge, UK, pp. 152–178. [NPR, MoS, SRC]
Ramaswamy, V., et al., 2001: Radiative forcing of climate change. In: Climate Change 2001: The Scientific Basis. Contribution of Working Group I to the Third Assessment Report of the Intergovernmental Panel on Climate Change [Houghton, J.T., et al. (eds.)]. Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, pp. 349–416. [NPR, MoS, SRC]
Ramaswamy, V., et al., 2006b: Anthropogenic and natural influences in the evolution of lower stratospheric cooling. Science, 311,1138–1141. [JoC, SRC]
Randall, C.E., R.M. Bevilacqua, J.D. Lumpe, and K.W. Hoppel, 2001: Validation of POAM III aerosols: Comparison to SAGE II and HALOE. J. Geophys. Res., 106,27525–27536. [JoC]
Randles, C.A., L.M. Russell, and V. Ramaswamy, 2004: Hygroscopic and optical properties of organic sea salt aerosol and consequences for climate forcing. Geophys. Res. Lett., 31,L16108, doi:10.1029/2004GL020628. [JoC, MoS, SRC]
Rasch, P.J., and J.E. Kristjánsson, 1998. A comparison of the CCM3 model climate using diagnosed and predicted condensate parametrizations. J. Clim., 11,1587–1614. [JoC, MoS]
Read, W.G., L. Froidevaux, and J.W. Waters, 1993: Microwave limb sounder measurements of stratospheric SO2 from the Mt. Pinatubo volcano. Geophys. Res. Lett., 20(12), 1299–1302. [JoC]
Reddy, M.S., and O. Boucher, 2004: A study of the global cycle of carbonaceous aerosols in the LMDZT general circulation model. J. Geophys. Res., 109,D14202, doi:10.1029/2003JD004048. [JoC, MoS, SRC]
Reddy, M.S., O. Boucher, Y. Balanski, and M. Schulz, 2005a: Aerosol optical depths and direct radiative perturbations by species and source type. Geophys. Res. Lett., 32,L12803, doi:10.1029/2004GL021743. [JoC, SRC]
Reddy, M.S., et al., 2005b: Estimates of global multicomponent aerosol optical depth and direct radiative perturbation in the Laboratoire de Météorologie Dynamique general circulation model. J. Geophys. Res., 110,D10S16, doi:10.1029/2004JD004757. [JoC, MoS]
Reid, J.S., et al., 1999: Use of the Angstrom exponent to estimate the variability of optical and physical properties of aging smoke particles in Brazil. J. Geophys. Res., 104(D22), 27473–27490. [JoC, MoS]
Reid, J.S., et al., 2003: Analysis of measurements of Saharan dust by airborne and ground-based remote sensing methods during the Puerto Rico Dust Experiment (PRIDE). J. Geophys. Res., 108(D19), 8586, doi:10.1029/2002JD002493. [JoC]
Reimann, S., et al., 2005: Low methyl chloroform emissions inferred from long-term atmospheric measurements. Nature, 433,506–508, doi:10.1038/nature03220. [JoC]
Remer, L.A., and Y.J. Kaufman, 2006: Aerosol direct radiative effect at the top of the atmosphere over cloud free oceans derived from four years of MODIS data. Atmos. Chem. Phys., 6,237–253. Clean
Remer, L.A., et al., 2002: Validation of MODIS aerosol retrieval over ocean. Geophys. Res. Lett., 29(12), doi:10.1029/2001GL013204. [JoC]
Remer, L.A., et al., 2005: The MODIS aerosol algorithm, products, and validation. J. Atmos. Sci., 62,947–973. [JoC]
Richards, J.F, 1990: Land transformation. In: The Earth as Transformed by Human Action [Turner, B.L. II, et al. (eds.)]. Cambridge University Press, New York, NY, pp. 163–178. [NPR]
Richter, A., et al., 2005: Increase in tropospheric nitrogen dioxide over China observed from space. Nature, 437,129–132. [JoC]
Rigozo, N.R., E. Echer, L.E.A. Vieira, and D.J.R. Nordemann, 2001: Reconstruction of Wolf sunspot numbers on the basis of spectral characteristics and estimates of associated radio flux and solar wind parameters for the last millennium. Sol. Phys., 203,179–191. [MoS]
Rind, D., J. Perlwitz, and P. Lonergan, 2005: AO/NAO response to climate change: 1. Respective influences of stratospheric and tropospheric climate changes. J. Geophys. Res., 110,D12107, doi:10.1029/2004JD005103. [JoC, SRC]
Rissler, J., et al., 2004: Physical properties of the sub-micrometer aerosol over the Amazon rain forest during the wet-to-dry season transition – Comparison of modeled and measured CCN concentrations. Atmos. Chem. Phys., 4,2119–2143. [MoS]
Roberts, D.L., and A. Jones, 2004: Climate sensitivity to black carbon aerosol from fossil fuel combustion. J. Geophys. Res., 109,D16202, doi:10.1029/2004JD004676. [JoC, ARC]
Robles-Gonzalez, C., J.P. Veefkind, and G. de Leeuw, 2000: Mean aerosol optical depth over Europe in August 1997 derived from ATSR-2 data. Geophys. Res. Lett. 27(7), 955–959. [JoC]
Robson, J.I., et al., 2006: Revised IR spectrum, radiative efficiency and global warming potential of nitrogen trifluoride. Geophys. Res. Lett., 33,L10817, doi:10.1029/2006GL026210. [JoC]
Rockmann, T., J. Grooss, and R. Muller, 2004: The impact of anthropogenic chlorine emissions, stratospheric ozone change and chemical feedbacks on stratospheric water. Atmos. Chem. Phys., 4,693–699. Clean
Rosenfeld, D., and G. Feingold, 2003: Explanation of discrepancies among satellite observations of the aerosol indirect effects. Geophys. Res. Lett., 30(14), 1776, doi:10.1029/2003GL017684. [JoC]
Rosenfeld, D., R. Lahav, A. Khain, and M. Pinsky, 2002: The role of sea spray in cleansing air pollution over the ocean via cloud processes. Science, 297,1667–1670. [JoC]
Ross, K.E., et al., 2003: Spatial and seasonal variations in CCN distribution and the aerosol-CCN relationship over southern Africa. J. Geophys. Res., 108(D13), 8481, doi:10.1029/2002JD002384. [JoC]
Rotstayn, L.D., 1997: A physically based scheme for the treatment of stratiform clouds and precipitation in large-scale models. I: Description and evaluation of the microphysical processes. Q. J. R. Meteorol. Soc., 123, 1227–1282. [JoC, MoS, ARC]
Rotstayn, L.D., and J.E. Penner, 2001: Indirect aerosol forcing, quasi forcing, and climate response. J. Clim., 14,2960–2975. [JoC, MoS, ARC]
Rotstayn, L.D., and Y. Liu, 2003: Sensitivity of the first indirect aerosol effect to an increase of the cloud droplet spectral dispersion with droplet number concentration. J. Clim., 16,3476–3481. [JoC, ARC]
Rotstayn, L.D., B.F. Ryan, and J. Katzfey, 2000: A scheme for calculation of the liquid fraction in mixed-phase clouds in large scale models. Mon. Weather Rev., 128,1070–1088. [JoC, MoS, ARC]
Rottman, G., 2005: The SORCE Mission. Solar Phys., 230,7–25. Clean
Rozanov, E.V., et al., 2002: Climate/chemistry effect of the Pinatubo volcanic eruption simulated by the UIUC stratosphere/troposphere GCM with interactive photochemistry. J. Geophys. Res., 107,4594, doi:10.1029/2001JD000974. [JoC, MoS]
Rozanov, E.V., et al., 2004: Atmospheric response to the observed increase of solar UV radiation from solar minimum to solar maximum simulated by the University of Illinois at Urbana-Champaign climate-chemistry model. J. Geophys. Res., 109,D01110, doi:10.1029/2003JD003796. [JoC, MoS]
Russell, P.B., and J. Heintzenberg, 2000: An overview of the ACE-2 clear sky column closure experiment (CLEARCOLUMN). Tellus, 52B,463–483, doi:10.1034/j.1600-0889.2000.00013.x. [JoC]
Russell, P.B., P.V. Hobbs, and L.L. Stowe, 1999: Aerosol properties and radiative effects in the United States East Coast haze plume: An overview of the Tropospheric Aerosol Radiative Forcing Observational Experiment (TARFOX). J. Geophys. Res., 104(D2), 2213–2222. [JoC, MoS]
Rypdal, K., et al., 2005: Tropospheric ozone and aerosols in climate agreements: scientific and political challenges. Environ. Sci. Policy, 8,29–43. Clean
Salby, M., and P. Callaghan, 2004: Evidence of the solar cycle in the general circulation of the stratosphere. J. Clim., 17,34–46. [JoC]
Santer, B.D., et al., 2004: Identification of anthropogenic climate change using a second-generation reanalysis. J. Geophys. Res., 109,D21104, doi:10.1029/2004JD005075. [PoC, JoC, ARC]
Sato, M., J.E. Hansen, M.P. McCormick, and J.B. Pollack, 1993: Stratospheric aerosol optical depths, 1850-1990. J. Geophys. Res., 98(D12), 22987–22994. [JoC, ARC]
Sato, M., et al., 2003: Global atmospheric black carbon inferred from AERONET. Proc. Natl. Acad. Sci. U.S.A., 100,6319–6324. [JoC]
Sausen, R., and U. Schumann, 2000: Estimates of the climate response to aircraft CO2 and NO(x) emissions scenarios. Clim. Change, 44,27–58. [JoC, MoS, SRC]
Sausen, R., K. Gierens, M. Ponater, and U. Schumann, 1998: A diagnostic study of the global distribution of contrails part I: Present day climate. Theor. Appl. Climatol., 61,127–141. [SRC]
Sausen, R., et al., 2005: Aviation radiative forcing in 2000: An update on IPCC (1999). Meteorol. Z., 14,1–7. [MoS, SRC]
Schaaf, C.B., et al., 2002: First operational BRDF, albedo nadir reflectance products from MODIS. Remote Sens. Environ., 83,135–148. Clean
Schaap, M., et al., 2004: Secondary inorganic aerosol simulations for Europe with special attention to nitrate. Atmos. Chem. Phys., 4, 857–874. [MoS]
Schatten, K.H., and J.A. Orosz, 1990: Solar constant secular changes. Sol. Phys., 125,179–184. Clean
Schmidt, G.A., et al., 2005: Present day atmospheric simulations using GISS ModelE: Comparison to in situ, satellite and reanalysis data. J. Clim., 19,153–192. [PoC, JoC, MoS, ARC]
Schnaiter, M., et al., 2003: UV-VIS-NIR spectral optical properties of soot and soot-containing aerosols. J. Aerosol Sci., 34(10), 1421–1444. Clean
Schoeberl, M., A. Douglass, Z. Zhu, and S. Pawson, 2003: A comparison of the lower stratospheric age spectra derived from a general circulation model and two data assimilation systems. J. Geophys. Res., 108,L4113, doi:10.1029/2002JD002652. [JoC, MoS]
Scholes, M., and M.O. Andreae, 2000: Biogenic and pyrogenic emissions from Africa and their impact on the global atmosphere. Ambio, 29,23–29. [JoC]
Schulz, M., S. Kinne, C. Textor, and S. Guibert, 2004: AeroCom Aerosol Model Intercomparison.http://nansen.ipsl.jussieu.fr/AEROCOM/. [NPR, MoS, SRC]
Schulz, M., et al., 2006: Radiative forcing by aerosols as derived from the AeroCom present-day and pre-industrial simulations. Atmos. Chem. Phys. Discuss., 6,5095–5136. [MoS, SRC]
Schumann, U., 2005: Formation, properties, and climatic effects of contrails. Comptes Rendus Physique, 6,549–565. Clean
Schuster, G.L., O. Dubovik, B.N. Holben, and E.E. Clothiaux, 2005: Inferring black carbon content and specific absorption from Aerosol Robotic Network (AERONET) aerosol retrievals. J. Geophys. Res., 110,D10S17, doi:10.1029/2004JD004548. [JoC]
Schwartz, S.E., and M.O. Andreae, 1996: Uncertainty in climate change caused by aerosols. Science, 272,1121–1122. [JoC, ARC]
Schwartz, S.E., D.W. Harshvardhan, and C.M. Benkovitz, 2002: Influence of anthropogenic aerosol on cloud optical depth and albedo shown by satellite measurements and chemical transport modeling. Proc. Natl. Acad. Sci. U.S.A., 99,1784–1789. [JoC, MoS, ARC]
Sekiguchi, M., et al., 2003: A study of the direct and indirect effects of aerosols using global satellite datasets of aerosol and cloud parameters. J. Geophys. Res., 108(D22), 4699, doi:10.1029/2002JD003359. [JoC]
Sellers, P.J., et al., 1996: Comparison of radiative and physiological effects of doubled atmospheric CO2 on climate. Science, 271,1402–1406. [JoC]
Shantz, N.C., W.R. Leaitch, and P. Caffrey, 2003: Effect of organics of low solubility on the growth rate of cloud droplets. J. Geophys. Res., 108(D5), doi:10.1029/2002JD002540. [JoC]
Sherwood, S., 2002: A microphysical connection among biomass burning, cumulus clouds, and stratospheric moisture. Science, 295,1272–1275. [JoC, ARC]
Shi, G.Y., et al., 2005: Sensitivity experiments on the effects of optical properties of dust aerosols on their radiative forcing under clear sky condition. J. Meteorol. Soc. Japan, 83A,333–346. [MoS]
Shindell, D.T., 2001: Climate and ozone response to increased stratospheric water vapor. Geophys. Res. Lett., 28,1551–1554. [JoC]
Shindell, D.T., and G. Faluvegi, 2002: An exploration of ozone changes and their radiative forcing prior to the chlorofluorocarbon era. Atmos. Chem. Phys., 2,363–374. [MoS]
Shindell, D.T., G. Faluvegi, and N. Bell, 2003a: Preindustrial-to-present day radiative forcing by tropospheric ozone from improved simulations with the GISS chemistry-climate GCM. Atmos. Chem. Phys., 3,1675–1702. [MoS]
Shindell, D.T., G.A. Schmidt, R.L. Miller, and M. Mann, 2003b: Volcanic and solar forcing of climate change during the preindustrial era. J. Clim., 16,4094–4107. [PoC, JoC, MoS, ARC]
Shindell, D.T., G.A. Schmidt, M. Mann, and G. Faluvegi, 2004: Dynamic winter climate response to large tropical volcanic eruptions since 1600. J. Geophys. Res., 109,D05104, doi:10.1029/2003JD004151. [PoC, JoC, ARC]
Shindell, D.T., G. Faluvegi, N. Bell, and G. Schmidt, 2005: An emissions-based view of climate forcing by methane and tropospheric ozone. Geophys. Res. Lett., 32,L04803, doi:10.1029/2004GL021900. [PoC, JoC, MoS, ARC]
Shine, K.P., 2005: Comment on ‘Contrails, cirrus, trends, and climate’. J. Clim., 18,2781–2782. [JoC]
Shine, K.P., J. Cook, E.J. Highwood, and M.M. Joshi, 2003: An alternative to radiative forcing for estimating the relative importance of climate change mechanisms. Geophys. Res. Lett., 30 (20), 2047, doi:10.1029/2003GL018141. [JoC, MoS]
Shine, K.P., T.K. Berntsen, J.S. Fuglestvedt, and R. Sausen, 2005a: Scientific issues in the design of metrics for inclusion of oxides of nitrogen in global climate agreements. Proc. Natl. Acad. Sci. U.S.A., 102,15768–15773. [JoC, SRC]
Shine, K.P., J.S. Fuglestvedt, K. Hailemariam, and N. Stuber, 2005b: Alternatives to the global warming potential for comparing climate impacts of emissions of greenhouse gases. Clim. Change, 68,281–302. [JoC, SRC]
Shine, K.P, et al., 2005c: Perfluorodecalin: global warming potential and first detection in the atmosphere. Atmos. Environ., 39,1759–1763. Clean
Simmonds, P.G., R. Derwent, A. Manning, and G. Spain, 2004: Significant growth in surface ozone at Mace Head, Ireland ,1987-2003. Atmos. Environ., 38,4769–4778. [SRC]
Simpson, I.J., D.R. Blake, F.S. Rowland, and T.Y. Chen, 2002: Implications of the recent fluctuations in the growth rate of tropospheric methane. Geophys. Res. Lett., 29(10), doi:10.1029/2001GL014521. [JoC]
Smith, C.A., J.D. Haigh, and R. Toumi, 2001: Radiative forcing due to trends in stratospheric water vapour. Geophys. Res. Lett., 28(1), 179–182. [JoC, MoS, ARC]
Smith, R.N.B., 1990: A scheme for predicting layer clouds and their water content in a general circulation model. Q. J. R. Meteorol. Soc., 116,435–460. [JoC, MoS]
Smith, S.J., and T.M.L. Wigley, 2000: Global warming potentials: 2. Accuracy. Clim. Change, 44,459–469. [PoC, JoC, ARC]
Smith, S.J., E. Conception, R. Andres, and J. Lurz, 2004: Historical Sulphur Dioxide Emissions 1850-2000: Methods and Results.Research Report No. PNNL-14537, Joint Global Change Research Institute, College Park, MD, 16 pp. [NPR, ARC]
Smith, W.L. Jr., et al., 2005: EOS terra aerosol and radiative flux validation: an overview of the Chesapeake Lighthouse and Aircraft Measurements for Satellites (CLAMS) experiment. J. Atmos. Sci., 62(4), 903–918, doi:10.1175/JAS3398.1. [JoC]
Sofia, S., and L.H. Li, 2001: Solar variability and climate. J. Geophys. Res., 106(A7), 12969–12974. [JoC]
Sokolov, A., 2006: Does model sensitivity to changes in CO2 provide a measure of sensitivity to the forcing of different nature. J. Clim., 19,3294–3306. [JoC, MoS, ARC]
Solanki, S.K., and M. Fligge, 1999: A reconstruction of total solar irradiance since 1700. Geophys. Res. Lett., 26(16), 2465–2468. [JoC, SRC]
Solanki, S.K., et al., 2004: Unusual activity of the Sun during recent decades compared to the previous 11,000 years. Nature, 431,1084–1087. [JoC, SRC]
Solomon, S., et al., 1996: The role of aerosol variations in anthropogenic ozone depletion at northern midlatitudes. J. Geophys. Res., 101(D3), 6713–6727. [PoC, JoC, ARC]
Solomon, S., et al., 2005: On the distribution and variability of ozone in the tropical upper troposphere: Implications for tropical deep convection and chemical-dynamical coupling. Geophys. Res. Lett., 32,L23813, doi:10.1029/2005GL024323. [PoC, JoC, ARC]
Soufflet, V., D. Tanre, A. Royer, and N.T. O’Neill, 1997: Remote sensing of aerosols over boreal forest and lake water from AVHRR data. Remote Sens. Environ., 60,22–34. Clean
Spahni, R., et al., 2005: Atmospheric methane and nitrous oxide of the late Pleistocene from Antarctic ice cores. Science, 310,1317–1321. [JoC, ARC]
Spinhirne, J.D., et al., 2005: Cloud and aerosol measurements from GLAS: overview and initial results. Geophys. Res. Lett., 32,L22S03, doi:10.1029/2005GL023507. [JoC]
Spruit, H., 2000: Theory of solar irradiance variations. Space Sci. Rev., 94,113–126. Clean
Steinbrecht, W., H. Claude, and P. Winkler, 2004a: Enhanced upper stratospheric ozone: sign of recovery or solar cycle effect? J. Geophys. Res., 109,D020308, doi:10.1029/2003JD004284. [JoC]
Steinbrecht, W., H. Claude, and P. Winkler, 2004b: Reply to comment by D. M. Cunnold et al. on “Enhanced upper stratospheric ozone: Sign of recovery or solar cycle effect?”. J. Geophys. Res., 109,D14306, doi:10.1029/2004JD004948. [JoC]
Stenchikov, G.L., et al., 1998: Radioactive forcing from the 1991 Mount Pinatubo volcanic eruption. J. Geophys. Res., 103(D12), 13837–13857. [JoC, MoS, SRC]
Stenchikov, G.L., et al., 2002: Arctic Oscillation response to the 1991 Mount Pinatubo eruption: effects of volcanic aerosols and ozone depletion. J. Geophys. Res., 107(D24), 4803, doi:10.1029/2002JD002090. [JoC, SRC]
Stenchikov, G., et al., 2004: Arctic Oscillation response to the 1991 Pinatubo eruption in the SKYHI GCM with a realistic quasi-biennial oscillation. J. Geophys. Res., 109,D03112, doi:10.1029/2003JD003699. [JoC, SRC]
Stenchikov, G., et al., 2006: Arctic Oscillation response to volcanic eruptions in the IPCC AR4 climate models. J. Geophys. Res., 111,D07107, doi:10.1029/2005JD006286. [JoC, MoS, SRC]
Stern, D.I., 2005: Global sulfur emissions from 1850 to 2000. Chemosphere, 58,163–175. Clean
Stevenson, D.S., et al., 2004: Radiative forcing from aircraft NOx emissions: mechanisms and seasonal dependence. J. Geophys. Res., 109,D17307, doi:10.1029/2004JD004759. [JoC, MoS, ARC]
Stier, P., et al., 2005: The aerosol-climate model ECHAM5-HAM. Atmos. Chem. Phys., 5,1125–1156. [MoS]
Stier, P., et al., 2006a: Impact of nonabsorbing anthropogenic aerosols on clear-sky atmospheric absorption. J. Geophys. Res., 111,D18201, doi:10.1029/2006JD007147. [JoC]
Stier, P., et al., 2006b: Emission-induced nonlinearities in the global aerosol system: results from the ECHAM5-HAM aerosol-climate model. J. Clim., 19,3845–3862. [JoC, MoS]
Stordal, F., et al., 2005: Is there a trend in cirrus cloud cover due to aircraft traffic? Atmos. Chem. Phys., 5,2155–2162. Clean
Stothers, R., 2001a: Major optical depth perturbations to the stratosphere from volcanic eruptions: Stellar extinction period, 1961-1978. J. Geophys. Res., 106(D3), 2993–3003. [JoC]
Stothers, R., 2001b: A chronology of annual mean radii of stratospheric aerosol from volcanic eruptions during the twentieth century as derived from ground-based spectral extinction measurements. J. Geophys. Res., 106(D23), 32043–32049. [JoC]
Stott P.A., G.S. Jones, and J.F.B. Mitchell, 2003: Do models underestimate the solar contribution to recent climate change? J. Clim., 16,4079–4093. [JoC, MoS]
Streets, D.G., et al., 2001: Black carbon emissions in China. Atmos. Environ., 35,4281–4296. Clean
Streets, D.G., et al., 2003: An inventory of gaseous and primary aerosol emissions in Asia in the year 2000. J. Geophys. Res., 108(D21), 8809, doi:10.1029/2002JD003093. [JoC]
Strugnell, N.C., W. Lucht, and C. Schaaf, 2001: A global albedo data set derived from AVHRR data for use in climate simulations. Geophys. Res. Lett., 28(1), 191–194. [JoC, MoS]
Stubenrauch, C.J., and U. Schumann, 2005: Impact of air traffic on cirrus coverage. Geophys. Res. Lett., 32,L14813, doi:10.1029/2005GL022707. [JoC]
Stuber, N., M. Ponater, and R. Sausen, 2001a: Is the climate sensitivity to ozone perturbations enhanced by stratospheric water vapor feedback? Geophys. Res. Lett., 28(15), 2887–2890. [JoC, SRC]
Stuber, N., R. Sausen, and M. Ponater, 2001b: Stratosphere adjusted radiative forcing calculations in a comprehensive climate model. Theor. Appl. Climatol., 68,125–135. [MoS, SRC]
Stuber, N., M. Ponater, and R. Sausen, 2005: Why radiative forcing might fail as a predictor of climate change. Clim. Dyn., 24,497–510. [JoC, MoS, SRC]
Sun, B., and R.S. Bradley, 2002: Solar influences on cosmic rays and cloud formation: a re-assessment. J. Geophys. Res., 107(D14), doi:10.1029/2001JD000560. [PoC, JoC]
Sundqvist, H., 1978: A parametrization scheme for non-convective condensation including prediction of cloud water content. Q. J. R. Meteorol. Soc., 104,677–690. [JoC, MoS]
Sundqvist, H., E. Berge, and J.E. Kristjánsson, 1989: Condensation and cloud parametrization studies with a mesoscale numerical weather prediction model. Mon. Weather Rev., 117,1641–1657. [JoC, MoS]
Suzuki, K., et al., 2004: A study of the aerosol effect on a cloud field with simultaneous use of GCM modeling and satellite observation. J. Atmos. Sci., 61,179–194. [JoC, MoS]
Svalgaard, L., E.W. Cliver, and P. Le Sager, 2004: IHV: A new long-term geomagnetic index. Adv. Space Res., 34,436–439. Clean
Swap, R.J., et al., 2002: The Southern African Regional Science Initiative (SAFARI 2000): overview of the dry season field campaign. S. Afr. J. Sci., 98,125–130. Clean
Swap, R.J., et al., 2003: Africa burning: a thematic analysis of the Southern African Regional Science Initiative (SAFARI 2000). J. Geophys. Res., 108(D13), 8465, doi:10.1029/2003JD003747. [JoC]
Tabazadeh, A., et al., 2002: Arctic “ozone hole” in cold volcanic stratosphere. Proc. Natl. Acad. Sci. U.S.A., 99,2609–2612. [JoC]
Takemura, T., T. Nakajima, T. Nozawa, and K. Aoki, 2001: Simulation of future aerosol distribution, radiative forcing, and long-range transport in East Asia. J. Meteorol. Soc. Japan, 2,79, 1139–1155. [MoS, SRC]
Takemura, T., et al., 2000: Global three-dimensional simulation of aerosol optical thickness distribution of various origins. J. Geophys. Res., 105(D14), 17853–17874. [JoC, MoS, SRC]
Takemura, T., et al., 2005: Simulation of climate response to aerosol direct and indirect effects with aerosol transport-radiation model. J. Geophys. Res., 110,D02202, doi:10.1029/2004JD005029. [JoC, MoS, SRC]
Tang, I.N., 1997: Thermodynamic and optical properties of mixed-salt aerosols of atmospheric importance. J. Geophys. Res., 102(D2), 1883–1893. [JoC]
Tang, I.N., K.H. Fung, D.G. Imre, and H.R. Munkelwitz, 1995: Phase transformation and metastability of hygroscopic microparticles. Aerosol Sci. Tech., 23,443. Clean
Tanré, D., Y.J. Kaufman, M. Herman, and S. Mattoo, 1997: Remote sensing of aerosol properties over oceans using the MODIS/EOS spectral radiances. J. Geophys. Res., 102(D14), 16971–16988. [JoC]
Tanré, D., et al., 2003: Measurement and modeling of the Saharan dust radiative impact: overview of the SaHAran Dust Experiment (SHADE). J. Geophys. Res., 108(D18), doi:10.1029/2002JD003273. [JoC, MoS]
Tarasick, D.W., et al., 2005: Changes in the vertical distribution of ozone over Canada from ozonesondes: 1980-2001. J. Geophys. Res., 110,D02304, doi:10.1029/2004JD004643. [JoC]
Tegen, I., and I. Fung, 1995: Contribution to the atmospheric mineral aerosol load from land surface modification. J. Geophys. Res., 100,18707–18726. [JoC, ARC]
Tegen, I., A.A. Lacis, and I. Fung, 1996: The influence on climate forcing of mineral aerosols from disturbed soils. Nature, 380,419–421. [JoC, MoS, ARC]
Tegen, I., M. Werner, S.P. Harrison, and K.E. Kohfeld, 2004: Relative importance of climate and land use in determining present and future global soil dust emission. Geophys. Res. Lett., 31,L05105, doi:10.1029/2003GL019216. [JoC, MoS]
Tegen, I., M. Werner, S.P. Harrison, and K.E. Kohfeld, 2005: Reply to comment by N. M. Mahowald et al. on “Relative importance of climate and land use in determining present and future global soil dust emission”. Geophys. Res. Lett., 32,doi:10.1029/2004GL021560. [NPR, JoC, MoS]
Tett, S.F.B., et al., 2002: Estimation of natural and anthropogenic contributions to twentieth century temperature change. J. Geophys. Res., 107(D16), 4306, doi:10.1029/2000JD000028. [JoC, MoS, ARC]
Textor, C., et al., 2006: AeroCom: The status quo of global aerosol modelling. Atmos. Chem. Phys., 6,1777–1813. [MoS, SRC]
Tie, X.X., G.P. Brasseur, B. Breiglib, and C. Granier, 1994: Two dimensional simulation of Pinatubo aerosol and its effect on stratospheric chemistry. J. Geophys. Res., 99(D10), 20545–20562. [JoC, MoS, ARC]
Thomason, L., and T. Peter, 2006: Assessment of Stratospheric Aerosol Properties (ASAP): Report on the Assessment Kick-Off Workshop, Paris, France, 4-6 November 2001.SPARC Report No. 4, WCRP-124, WMO/TD No. 1295, http://www.aero.jussieu.fr/~sparc/News18/18_Thomason.html. [NPR]
Thompson, A.M., et al., 2001: Tropical tropospheric ozone and biomass burning. Science, 291,2128–2132. [JoC]
Thompson, T.M., et al., 2004: Halocarbons and other atmospheric trace species. In: Climate Monitoring and Diagnostics Laboratory, Summary Report No. 27 [Schnell, R.C., A.-M. Buggle, and R.M. Rosson (eds.)]. NOAA CMDL, Boulder, CO, pp. 115–135. [NPR]
Timmreck, C., and M. Schulz, 2004: Significant dust simulation differences in nudged and climatological operation mode of the AGCM ECHAM. J. Geophys. Res., 109,D13202, doi:10.1029/2003JD004381. [JoC, MoS, SRC]
Timmreck, C., H.-F. Graf, and B. Steil, 2003: Aerosol chemistry interactions after the Mt. Pinatubo Eruption. In: Volcanism and the Earth’s Atmosphere [Robock, A., and C. Oppenheimer (eds.)]. Geophysical Monograph 139, American Geophysical Union, Washington, DC, pp. 227–236. [NPR]
Tol, R.S.J., 2002: Estimates of the damage costs of climate change, Part II. Dynamic estimates. Environ. Resour. Econ., 21,135–160. [MoS]
Torres, O., et al., 2002: A long-term record of aerosol optical depth from TOMS: Observations and comparison to AERONET measurements. J. Atmos. Sci., 59,398–413. [JoC]
Travis, D.J., A.M. Carleton, and R.G. Lauritsen, 2002: Contrails reduce daily temperature range. Nature, 418,601–602. [JoC]
Travis, D.J., A.M. Carleton, and R.G. Lauritsen, 2004: Regional variations in U.S. diurnal temperature range for the 11-14 September 2001 aircraft groundings: evidence of jet contrail influence on climate. J. Clim., 17,1123–1134. [JoC]
Tripoli, G.J., and W.R. Cotton, 1980: A numerical investigation of several factors contributing to the observed variable intensity of deep convection over South Florida. J. Appl. Meteorol., 19,1037–1063. [JoC]
Twohy, C.H., et al., 2005: Evaluation of the aerosol indirect effect in marine stratocumulus clouds: droplet number, size, liquid water path, and radiative impact. J. Geophys.Res., 110,D08203, doi:10.1029/2004JD005116. [JoC]
Twomey, S.A., 1977: The influence of pollution on the shortwave albedo of clouds. J. Atmos. Sci., 34,1149–1152. [JoC]
Udelhofen, P.M., and R.D. Cess, 2001: Cloud cover variations over the United States: An influence of cosmic rays or solar variability? Geophys. Res. Lett., 28(13), 2617–2620. [JoC]
Usoskin, I.G., et al., 2004: Latitudinal dependence of low cloud amount on cosmic ray induced ionization. Geophys. Res. Lett., 31,L16109, doi:10.1029/2004GL019507. [JoC]
Van Aardenne, J.A., et al., 2001: A 1 x 1 degree resolution dataset of historical anthropogenic trace gas emissions for the period 1890-1990. Global Biogeochem. Cycles, 15,909–928. [JoC]
van der Werf, et al., 2004: Continental-scale partitioning of fire emissions during the 1997 to 2001 El Nino/La Nina period. Science, 303,73–76. [JoC]
Van Dorland, R., F.J. Dentener, and J. Lelieveld, 1997: Radiative forcing due to tropospheric ozone and sulfate aerosols. J. Geophys. Res., 102(D23), 28079–28100. [JoC, MoS, SRC]
van Loon, H., and D.J. Shea, 2000: The global 11-year solar signal in July-August. Geophys. Res. Lett., 27(18), 2965–2968. [JoC, ARC]
Veefkind, J.P., G. de Leeuw, and P.A. Durkee, 1998: Retrieval of aerosol optical depth over land using two-angle view satellite radiometry. Geophys. Res. Lett. 25(16), 3135–3138. [JoC]
Velders, et al., 2005: Chemical and radiative effects of halocarbons and their replacement compounds. In: Special Report on Safeguarding the Ozone Layer and the Global Climate System: Issues Related to Hydrofluorocarbons and Perfluorocarbons [Metz, B., et al. (eds.)]. Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, pp. 133–180. [NPR]
Vestreng, V., M. Adams, and J. Goodwin, 2004: Inventory Review 2004: Emission Data Reported to CLRTAP and the NEC Directive.EMEP/EEA Joint Review Report, Norwegian Meteorological Institute, Norway, 120 pp. [NPR]
von Hoyningen-Huene, W., M. Freitag, and J.B. Burrows, 2003: Retrieval of aerosol optical thickness over land surfaces from top-of-atmosphere radiance. J. Geophys. Res., 108(D9), 4260, doi:10.1029/2001JD002018. [JoC]
Walter, B.P., M. Heimann, and E. Matthews, 2001a: Modeling modern methane emissions from natural wetlands 2. Interannual variations 1982-1993. J. Geophys. Res., 106(D24), 34207–34220. [JoC, MoS, ARC]
Walter, B.P., M. Heimann, and E. Matthews, 2001b: Modeling modern methane emissions from natural wetlands 1. Model description and results. J. Geophys. Res., 106(D24), 34189–34206. [JoC, MoS, ARC]
Wang, C., 2004: A modeling study on the climate impacts of black carbon aerosols. J. Geophys. Res., 109,D03106, doi:10.1029/2003JD004084. [JoC, MoS]
Wang, H.J., et al., 2002: Assessment of SAGE version 6.1 ozone data quality. J. Geophys. Res., 107(D23), 4691, doi:10.1029/2002JD002418. [JoC]
Wang, M.H., K.D. Knobelspiesse, and C.R. McClain, 2005: Study of the Sea-Viewing Wide Field-of-View Sensor (SeaWiFS) aerosol optical property data over ocean in combination with the ocean color products. J. Geophys. Res., 110,D10S06, doi:10.1029/2004JD004950. [JoC, ARC]
Wang, W.-C., M.P. Dudek, and X.-Z. Liang, 1992: Inadequacy of effective CO2 as a proxy in assessing the regional climate change due to other radiatively active gases. Geophys. Res. Lett., 19,1375–1378. [JoC]
Wang, W.-C., M.P. Dudek, X.-Z. Liang, and J.T. Kiehl, 1991: Inadequacy of effective CO2 as a proxy in simulating the greenhouse effect of other radiatively active gases. Nature, 350,573–577. [JoC, MoS]
Wang, Y.M., J.L. Lean, and N.R. Sheeley, 2005: Modeling the sun’s magnetic field and irradiance since 1713. Astrophys. J., 625,522–538. [MoS, SRC]
Warner, J., and S.A. Twomey, 1967: The production and cloud nuclei by cane fires and the effect on cloud droplet concentration. J. Atmos. Sci., 24,704–706. [JoC]
Warwick, N.J., et al., 2002: The impact of meteorology on the interannual growth rate of atmospheric methane. Geophys. Res. Lett., 29(26), doi:10.1029/2002/GL015282. [JoC]
Weatherhead, E.C., and S. B. Andersen, 2006: The search for signs of recovery of the ozone layer. Nature, 441,39–45. [JoC]
Welton, E.J., J.R. Campbell, J.D. Spinhirne, and V.S. Scott, 2001: Global monitoring of clouds and aerosols using a network of micro-pulse lidar systems. In: Lidar Remote Sensing for Industry and Environmental Monitoring [Singh, U.N., T. Itabe, and N. Sugimoto (eds.)]. SPIE, Bellingham, WA, pp. 151–158. [NPR]
Wennberg, P.O., S. Peacock, J.T. Randerson, and R. Bleck, 2004: Recent changes in the air-sea gas exchange of methyl chloroform. Geophys. Res. Lett., 31,L16112, doi:10.1029/2004GL020476. [JoC]
Westerling, A.L., H.G. Hidalgo, D.R. Cayan, and T.W. Swetnam, 2006: Warming and earlier spring increase western U.S. forest wildfire activity. Science, 313,940–943. [JoC, ARC]
White, W.B., M.D. Dettinger, and D.R. Cayan, 2003: Sources of global warming of the upper ocean on decadal period scales. J. Geophys. Res., 108(C8), doi:10.1029/2002JC001396. [JoC, ARC]
Wild, O., M.J. Prather, and H. Akimoto, 2001: Indirect long-term global radiative cooling from NOx emissions. Geophys. Res. Lett., 28(9), 1719–1722. [JoC, ARC]
Williams, K.D., C.A. Senior, and J.F.B. Mitchell, 2001a: Transient climate change in the Hadley Centre models: the role of physical processes. J. Clim., 14,2659–2674. [JoC, MoS, ARC]
Williams, K.D., et al., 2001b: The response of the climate system to the indirect effects of anthropogenic sulfate aerosols. Clim. Dyn., 17,846–856. [JoC]
Willson, R.C., and A.V. Mordvinov, 2003: Secular total solar irradiance trend during solar cycles 21-23. Geophys. Res. Lett., 30(5), 3–6. [JoC]
Wilson, D.R., and S.P. Ballard, 1999: A microphysically based precipitation scheme for the UK Meteorological Office Unified Model. Q. J. R. Meteorol. Soc., 125,1607–1636. [JoC, MoS]
Wilson, M.F., and A. Henderson-Sellers, 1985: A global archive of land cover and soils data for use in general-circulation climate models. J. Climatol., 5,119–143. [MoS, ARC]
WMO, 1986: Atmospheric Ozone 1985.Global Ozone Research and Monitoring Project Report No.16, World Meteorological Organisation, Geneva, Volume 3. [NPR]
WMO, 2003: Scientific Assessment of Ozone Depletion: 2002.Global Ozone Research and Monitoring Project Report No. 47, World Meteorological Organization, Geneva, 498 pp. [NPR]
Wong, J., and Z. Li, 2002: Retrieval of optical depth for heavy smoke aerosol plumes: uncertainties and sensitivities to the optical properties. J. Atmos. Sci., 59,250–261. [JoC]
Wong, S., et al., 2004: A global climate-chemistry model study of present-day tropospheric chemistry and radiative forcing from changes in troposphericO 3 since the preindustrial period. J. Geophys. Res., 109,D11309, doi:10.1029/2003JD003998. [JoC, MoS]
Woods, T.N., et al., 1996: Validation of the UARS solar ultraviolet irradiances: comparison with the ATLAS 1 and 2 measurements. J. Geophys. Res., 101(D6), 9541–9569. [JoC]
Xiong, J.Q., et al., 1998: Influence of organic films on the hygroscopicity of ultrafine sulfuric acid aerosol. Environ. Sci. Technol., 32,3536–3541. Clean
Xue, H., and G. Feingold, 2006: large eddy simulations of trade-wind cumuli: investigation of aerosol indirect effects. J. Atmos. Sci., 63,1605–1622. [JoC, MoS]
Yang, F., and M. Schlesinger, 2001: Identification and separation of Mount Pinatubo and El Nino-Southern Oscillation land surface temperature anomalies. J. Geophys. Res., 106(D14), 14757–14770. [JoC]
Yang, F., and M. Schlesinger, 2002: On the surface and atmospheric temperature changes following the 1991 Pinatubo volcanic eruption: a GCM study. J. Geophys. Res., 107(D8), doi:10.1029/2001JD000373. [JoC]
Yokouchi, et al., 2005: Estimates of ratios of anthropogenic halocarbon emissions from Japan based on aircraft monitoring over Sagami Bay, Japan. J. Geophys. Res., 110,D06301, doi:10.1029/2004JD005320. [JoC, MoS]
Yoshioka, M., N. Mahowald, J.L. Dufresne, and C. Luo, 2005: Simulation of absorbing aerosol indices for African dust. J. Geophys. Res., 110,D18S17, doi:10.1029/2004JD005276. [JoC, MoS, ARC]
Yu, H., et al., 2003: Annual cycle of global distributions of aerosol optical depth from integration of MODIS retrievals and GOCART model simulations. J. Geophys. Res., 108(D3), 4128, doi:10.1029/2002JD002717. [JoC, MoS]
Yu, H., et al., 2006: A review of measurement-based assessments of the aerosol direct radiative effect and forcing. Atmos. Chem. Phys., 6,613–666. [MoS]
Yvon-Lewis, S.A., and J.H. Butler, 2002: Effect of oceanic uptake on atmospheric lifetimes of selected trace gases. J. Geophys. Res., 107(D20), 4414, doi:10.1029/2001JD001267. [JoC]
Zender, C.S., 2004: Quantifying mineral dust mass budgets: terminology, constraints, and current estimates. Eos, 85,509–512. [JoC, MoS]
Zerefos, C.S., et al., 2003: Evidence of impact of aviation on cirrus cloud formation. Atmos. Chem. Phys., 3,1633–1644. Clean
Zhang, J., and S. Christopher, 2003: Longwave radiative forcing of Saharan dust aerosols estimated from MODIS, MISR and CERES observations on Terra. Geophys. Res. Lett., 30(23), doi:10.1029/2003GL018479. [JoC, MoS]
Zhang, J., S.A. Christopher, L.A. Remer, and Y.J. Kaufman, 2005: Shortwave aerosol radiative forcing over cloud-free oceans from Terra: 2. Seasonal and global distributions. J. Geophys. Res., 110,D10S24, doi:10.1029/2004JD005009. [JoC, MoS]
Zhang, J.P., Z. Yang, D.J. Wang, and X.B. Zhang, 2002: Climate change and causes in the Yuanmou dry-hot valley of Yunnan China. J. Arid Environ., 51,153–162. [ARC]
Zhao, M., A. Pitman, and T.N. Chase, 2001: The impacts of land cover change on the atmospheric circulation. Clim. Dyn., 17,467–477. [JoC, ARC]
Zhao, T.X.P., I. Laszlo, P. Minnis, and L. Remer, 2005: Comparison and analysis of two aerosol retrievals over the ocean in the terra/clouds and the earth’s radiant energy system: moderate resolution imaging spectroradiometer single scanner footprint data: 1. Global evaluation. J. Geophys. Res., 110,D21208, doi:10.1029/2005JD005851. [JoC]
Zhou, L.M., et al., 2001: Variations in northern vegetation activity inferred from satellite data of vegetation index during 1981 to 1999. J. Geophys. Res., 106(D17), 20069–20083. [JoC, ARC]
Zhou, M., et al., 2005: A normalized description of the direct effect of key aerosol types on solar radiation as estimated from aerosol robotic network aerosols and moderate resolution imaging spectroradiometer albedos. J. Geophys. Res., 110,D19202, doi:10.1029/2005JD005909.[JoC, MoS]
| Tag | Explanation | Occurrences | Percentages |
|---|---|---|---|
| PoC |
Person of Concern Key individual involved in CRU emails as defined in this spreadsheet. |
19 | 2% |
| JoC |
Journal of Concern A Journal which has published articles by one or more PoCs (Person of Concern) |
585 | 77% |
| MoS |
Model or Simulation Reference appears to be a model or simulation, not observation or experiment. |
262 | 34% |
| NPR |
Non Peer Reviewed Reference has no Journal or no Volume or no Pages or it has Editors. |
54 | 7% |
| SRC |
Self Reference Concern Author of a chapter containing references to own work. |
165 | 21% |
| ARC |
Paper authored or co-authored by person who is also in list of Authors of another chapter. |
113 | 14% |
| 2007 |
Paper dated 2007, when IPCC policy stated cutoff was December 2005 |
6 | 0.7% |
| Clean |
The reference was probably peer reviewed. |
68 | 8% |
NOTE: In the Summary of Reviewer Comments,
Accepted includes only those responses that were unambiguously Accepted.
There are other categories of responses that we have not yet quantified
due to inconsistencies of usage.
| Reviewer | Total Comments | Accepted | IPCC Roles | Papers |
|---|---|---|---|---|
| Gray, Vincent | 171 | 29 | 0 | 0 |
| Gillett, Nathan | 99 | 54 | 0 | 0 |
| Govt. of United States of America | 71 | 19 | 0 | 0 |
| Kristjansson, Je | 50 | 29 | 0 | 0 |
| Peter, T | 44 | 30 | 0 | 3 |
| Govt. of Finland | 35 | 18 | 0 | 0 |
| Govt. of Australia | 32 | 25 | 0 | 0 |
| Sun, Junying | 30 | 22 | 0 | 0 |
| Penner, JE | 28 | 9 | 2 | 21 |
| Harvey, D | 26 | 18 | 1 | 1 |
| Marcolli, Claudia | 25 | 12 | 0 | 0 |
| Shine, Keith | 25 | 14 | 0 | 0 |
| Govt. of Germany | 24 | 12 | 0 | 0 |
| Jacobson, Mark | 20 | 8 | 0 | 0 |
| Leck, C | 20 | 6 | 1 | 4 |
| Boucher, O | 19 | 7 | 1 | 18 |
| Manton, Michael | 18 | 5 | 0 | 0 |
| Commission, European | 17 | 8 | 0 | 0 |
| Feingold, G | 15 | 12 | 0 | 13 |
| Yu, H | 14 | 6 | 0 | 2 |
| Tegen, I | 14 | 6 | 0 | 12 |
| Hare, William | 14 | 9 | 0 | 0 |
| Jones, Gs | 14 | 9 | 0 | 0 |
| Govt. of Belgium | 13 | 8 | 0 | 0 |
| Simmons, AJ | 13 | 7 | 1 | 3 |
| Collins, William | 13 | 10 | 0 | 0 |
| Muller, R | 12 | 6 | 0 | 2 |
| Roscoe, Hk | 12 | 8 | 0 | 0 |
| Lassey, KR | 12 | 7 | 2 | 1 |
| Millet, Dylan | 12 | 4 | 0 | 0 |
| Danilin, Michael | 11 | 7 | 0 | 0 |
| Rossi, Mj | 10 | 5 | 0 | 0 |
| Colombo, Tiziano | 9 | 5 | 0 | 0 |
| Owens, John | 9 | 7 | 0 | 0 |
| Haigh, JD | 8 | 5 | 1 | 6 |
| Ko, Malcolm | 8 | 3 | 0 | 0 |
| Daniel, Js | 8 | 2 | 0 | 0 |
| Enting, Ian | 7 | 6 | 0 | 0 |
| Murphy, Daniel | 6 | 4 | 0 | 0 |
| Fahey, Dw&D | 6 | 6 | 0 | 0 |
| Hamill, P | 6 | 5 | 0 | 1 |
| Martin, Scot | 6 | 0 | 0 | 0 |
| Weiss, RF | 6 | 2 | 1 | 5 |
| Jones, CD | 6 | 3 | 3 | 10 |
| Govt. of Norway | 6 | 1 | 0 | 0 |
| Bergamaschi, P | 5 | 3 | 0 | 2 |
| Prinn, RG | 5 | 2 | 1 | 13 |
| Bernstein, Lenny | 5 | 1 | 0 | 0 |
| Velders, Guus | 5 | 4 | 0 | 0 |
| Kueter, Jeff | 5 | 0 | 0 | 0 |
| Li, Z-X | 5 | 1 | 0 | 6 |
| Evans, Wf | 4 | 0 | 0 | 0 |
| Govt. of China | 4 | 3 | 0 | 0 |
| Siems, Steven | 4 | 2 | 0 | 0 |
| Solomon, SM | 4 | 2 | 2 | 11 |
| Baughcum, Steven | 4 | 2 | 0 | 0 |
| Held, IM | 4 | 3 | 1 | 8 |
| Benestad, RE | 4 | 2 | 1 | 7 |
| Govt. of Japan | 4 | 1 | 0 | 0 |
| Anderson, Wilmer | 4 | 4 | 0 | 0 |
| Mikami, M | 4 | 1 | 0 | 1 |
| Tsushima, Y | 3 | 3 | 0 | 2 |
| Manning, MR | 3 | 1 | 2 | 5 |
| Bates, TS | 3 | 0 | 0 | 3 |
| Maccracken, Michael | 3 | 1 | 0 | 0 |
| Garivait, Savitri | 3 | 0 | 0 | 0 |
| Govt. of Ireland | 2 | 0 | 0 | 0 |
| Zagoni, Miklos | 2 | 0 | 0 | 0 |
| Govt. of France | 2 | 0 | 0 | 0 |
| Tsutsumi, Yukitomo | 2 | 2 | 0 | 0 |
| Quere, Cl | 2 | 1 | 0 | 0 |
| Govt. of Spain | 2 | 1 | 0 | 0 |
| Govt. of Hungary | 2 | 0 | 0 | 0 |
| Guo, Xueliang | 2 | 1 | 0 | 0 |
| Soulen, Richard | 2 | 1 | 0 | 0 |
| Wurzler, S | 2 | 2 | 0 | 4 |
| Sievering, H | 2 | 0 | 0 | 2 |
| Shindell, Drew | 2 | 0 | 0 | 0 |
| Maki, Takashi | 2 | 0 | 0 | 0 |
| Otto-Bliesner, BL | 2 | 1 | 2 | 6 |
| Plattner, GK | 2 | 0 | 3 | 2 |
| Luterbacher, J | 1 | 0 | 1 | 6 |
| Lacis, AA | 1 | 0 | 0 | 3 |
| Govt. of United Kingdom | 1 | 0 | 0 | 0 |
| Stevens, B | 1 | 1 | 0 | 2 |
| Kheshgi, HS | 1 | 1 | 0 | 2 |
| Kandel, Robert | 1 | 0 | 0 | 0 |
| Govt. of Republic of Korea | 1 | 0 | 0 | 0 |
| Knutti, R | 1 | 0 | 5 | 9 |
| Mcintyre, Stephen | 1 | 0 | 0 | 0 |
| Backer, Hd | 1 | 0 | 0 | 0 |
| Tsu, Wg1 | 1 | 1 | 0 | 0 |
| Mckitrick, Ross | 1 | 0 | 0 | 0 |
| Trishchenko, Alexander | 1 | 0 | 0 | 0 |
| Wang, JH | 1 | 1 | 0 | 4 |
| Wolff, Eric | 1 | 1 | 0 | 0 |
| Govt. Of Finland | 1 | 1 | 0 | 0 |
| Giorgetta, Ma | 1 | 0 | 0 | 0 |
| Schmidt, GA | 1 | 0 | 1 | 14 |
| Totals | 1157 | 529 | 32 | 214 |
| Reviewer Type | Total Comments | Accepted | % Accepted |
|---|---|---|---|
| Govt | 200 | 89 | 44% |
| IPCC | 298 | 153 | 51% |
| Indep | 659 | 287 | 43% |
| Total | 1157 | 529 | 45% |
Main AR4 Index | Working Group WG1 Index | Table of Contents | Authors | Executive Summary | Annotated Text | References | Reviewer Comments